一种吗啡衍生物的合成方法及应用与流程
本发明涉及镇痛药领域的药物合成,尤其涉及吗啡衍生物的合成方法。
背景技术:
吗啡在体内代谢为吗啡-3-葡糖醛酸苷(m3g)和吗啡-6-葡糖醛酸苷(m6g),其中,m3g占代谢物的44%~45%,没有镇痛活性。m3g具有较大的极性,不易透过血脑屏障。然而,在制备m6g的过程中,有可能也生成了m3g,从而影响了m6g镇痛活性的发挥。
文献1bertoldberrang,etal,synthesisofmorphine-3-glucuronide,synthesiscommunications,5(3),231-236(1975),报道了一种合成m3g的合成方法。采用吗啡在氢氧化锂作用下,与糖环反应,再在氢氧化锂作用下水解,制得m3g。
然而,本发明人在重复该方法时,发现在氢氧化锂催化下,吗啡几乎不能和糖环发生反应,hplc显示几乎都是吗啡的峰,糖苷的峰几乎看不到;经氢氧化锂水解处理后,产物中也存在大量的吗啡,经纯化后不能得到较纯的m3g(m3g的hplc纯度约为65%,吗啡为35%)。因此,使用该方法并无法得到纯度较好的m3g,也不能作为标准品或对照品。
因此,需要开发一种制备m3g的新方法,经过简单的纯化后可得到比较纯的产物,作为标准品或对照品,为进行m6g的质量研究提供参考依据。
技术实现要素:
本发明人开发了一种合成m3g的方法,通过该方法合成的m3g,hplc纯度可以达到99.8%以上,可以作为标准品或对照品,用于m6g的定性或定量的质量研究。
m3g的结构如式i所示:
本发明的目的是提供一种吗啡-3-葡萄糖醛酸苷的合成方法。
本发明的另一个目的提供吗啡-3-葡萄糖醛酸苷作为标准品或对照品的应用,用于吗啡-6-葡萄糖醛酸苷的定性或定量研究。
在本发明的实施方案中,本发明提供了一种吗啡-3-葡萄糖醛酸苷的合成方法,包括如下步骤:
(1)吗啡在催化剂作用下,与酰氯r1-cl或酸酐r1-o-r1成酯,得式ii所示化合物;
(2)式ii所示化合物在催化剂作用下脱去3-位保护基得式iii所示化合物;
(3)式iii所示化合物与三氟乙酸酐或三氟甲磺酸酐反应得式iv所示化合物;
(4)式iv所示化合物在路易斯酸作用下与式v所示化合物反应得到式vi所示化合物;
(5)式vi所示化合物在碱作用下水解为吗啡-3-葡萄糖醛酸苷,即式i所示化合物;
其中,酰氯r1-cl或酸酐r1-o-r1、式ii至vi所示化合物中的r1为乙酰基、异丁酰基、特戊酰基、苯甲酰基、对甲基苯磺酰基、硝基苯甲酰基、ch2cl-co-、chcl2-co-、ccl3-co-、ch3o-co-co-、ch3o-co-、ch3ch2o-co-、或ch3ch2o-co-co-;
式v至vi所示化合物中的r2为乙酰基、异丁酰基、或特戊酰基;
式v至vi所示化合物中的r3为甲基、或乙基;
式v化合物中r4为乙酰氧基、异丁酰氧基、三氟乙酰氧基、三氟甲磺酰氧基、溴、或碘;
式iv化合物中r5为三氟乙酰基、或三氟甲磺酰基。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(1)所述的酰氯为乙酰氯、异丁酰氯、特戊酰氯、氯代乙酰氯、苯甲酰氯、对甲基苯磺酰氯、硝基苯甲酰氯、草酰氯单乙酯、草酰氯单甲酯、氯甲酸乙酯、或氯甲酸甲酯;所述的酸酐为乙酸酐、二氯醋酸酐、三氯乙酸酐、或苯甲酸酐。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(1)所述的催化剂为三乙胺、吡啶、氢化钠、吗啉、碳酸钾、或碳酸钠。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(2)所述的催化剂为盐酸羟氨、碳酸钠、碳酸钾、丁胺、氨气、苯肼、硼酸钠、或硼氢化钠。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(2)的反应是在质子有机溶剂中进行的,所述质子有机溶剂为甲醇、乙醇、异丙醇、或乙二醇。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(4)所述的路易斯酸为溴化铜、溴化亚铜、溴化锌、三氟化硼、三氯化铝、三甲基硅烷三氟甲磺酸酯、或碳酸银。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(4)所述的反应是在非质子有机溶剂中进行的,所述非质子有机溶剂为二氯甲烷、三氯甲烷、硝基甲烷、乙腈、苯、甲苯、或二甲苯。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(4)的反应温度为20~160℃。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(5)的反应是在低级烷醇中进行的,所述低级烷醇为甲醇、乙醇、或异丙醇。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(5)的反应温度为-50~0℃。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,步骤(5)所述的碱为氢氧化钠、氢氧化钾、叔丁醇钾、氢氧化钙、或氢氧化钡。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,在步骤(5)水解后,进一步地包括分离和纯化步骤,所述的分离和纯化包括选自重结晶、柱层析、制备型液相分离精制的任一种或其组合。这里,所述柱层析和制备型液相分离精制的条件为:流动相或洗脱剂为甲醇-水、乙腈-水、甲醇-乙腈-水的任一种或其组合,进行等度或梯度洗脱,收集产品流份,蒸干或冷冻干燥即得。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,式v所示化合物可以通过如下方式获得:
式v所示化合物(r2和r4为相同的保护基)的合成
在20l反应瓶中加入15l甲醇、乙醇6g氢氧化钠,搅拌溶解。开始分批加入2000gd-葡萄糖醛酸内酯,加料完毕后保持30℃反应8小时。45℃减压浓缩得到红褐色粘稠固体。向上步剩余物中加入2l吡啶来溶解并45℃加热溶解,然后把反应液转入到50l的反应釜中,接着向反应釜中加入10l二氯甲烷,降温冷却到0℃,开始滴加10l乙酰氯、乙酸酐、异丁酰氯或特戊酰氯,滴加过程中控制温度在15℃以下。滴加完毕以后,常温搅拌过夜。然后60℃回流反应7小时,停止反应。慢慢滴加4l甲醇,控制滴加过程中反应温度在20℃以下。再加入10l水,分出有机层。有机层依次用15l10%hcl,10l10%碳酸氢钠溶液,饱和氯化钠溶液洗涤。有机相45℃减压浓缩得到红褐色粘稠固体,用5l乙醇溶解,冷冻析晶。减压抽滤并用冷冻乙醇淋洗,60℃常压鼓风干燥,得到白色或类白色固体。收率85%左右。
或者,式v所示化合物(r2和r4为不同的保护基)可参照下列方法来合成:
例如,将r2和r4均为异丁酰基、r3为甲基的式v所示化合物20g溶于乙酸中,10g三氟乙酸酐,加入三氟化硼0.1ml,30~40℃反应,倒入冰水中,过滤,滤饼45~50℃减压干燥,得到r2为异丁酰基、r3为甲基、r4为三氟乙酰基的式v所示化合物20.5g,收率97.6%;
式v所示化合物(r2为乙酰基,r4为溴,r3为甲基)的合成
将r2和r4均为乙酰基、r3为甲基的式v所示化合物20g溶于10ml乙酸中,加入溴化氢乙酸溶液30ml,20~30℃反应,倒入冰水中,用50ml×3二氯甲烷萃取,用饱和碳酸氢钠溶液中和,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用甲醇重结晶,得式v所示化合物10.5g(r2为乙酰基、r3为甲基、r4为溴)。
在本发明的实施方案中,本发明提供的一种吗啡-3-葡萄糖醛酸苷的合成方法,其中,式iv所示化合物的合成方法
将式iii所示化合物0.1mol用乙腈或四氢呋喃溶解,控制反应温度在-5~5℃,滴加0.8~1.5当量的三氟甲磺酸酐或三氟乙酸酐,滴加完毕,自然升温至室温反应4~6小时,反应完毕,将反应体系倒入二氯甲烷或乙酸乙酯中,用碳酸氢钠溶液中和,有机相用无水硫酸镁干燥,过滤,减压浓缩,得式vi所示化合物。
另一方面,本发明提供了吗啡-3-葡萄糖醛酸苷作为标准品或对照品,用于药品质量研究的应用,特别是用于m6g等相关产品的质量研究的应用。
本发明合成方法得到的m3g,hplc的纯度可以达到99.8%,其质量远远高于目前文献报道的合成方法得到的m3g,完全可以作为标准品或杂质对照品用于有关产品的质量研究。
此外,在本发明合成方法的研究中,本发明令人惊奇地发现,式(iii)所示化合物中的3-酚羟基如果不用三氟乙酰基或三氟甲磺酰基进行保护,在进行糖苷化反应的过程中,由于3-酚羟基比6-醇羟基活泼,不能像制备m6g的糖苷化中间体那样始终保持着糖苷化反应,在合成m3g的糖苷化反应中,会有部分式(iii)所示化合物与糖环上的酯键发生酯交换反应,造成产物的分离困难。
术语定义如下:
乙酰基:ch3co-;三氟乙酰基:cf3co-;三氟甲磺酰基:cf3so2-;乙酰氧基:ch3coo-;三氟乙酰氧基:cf3coo-;三氟甲磺酰氧基:cf3so2-异丁酰氧基:
异丁酰基:
附图说明
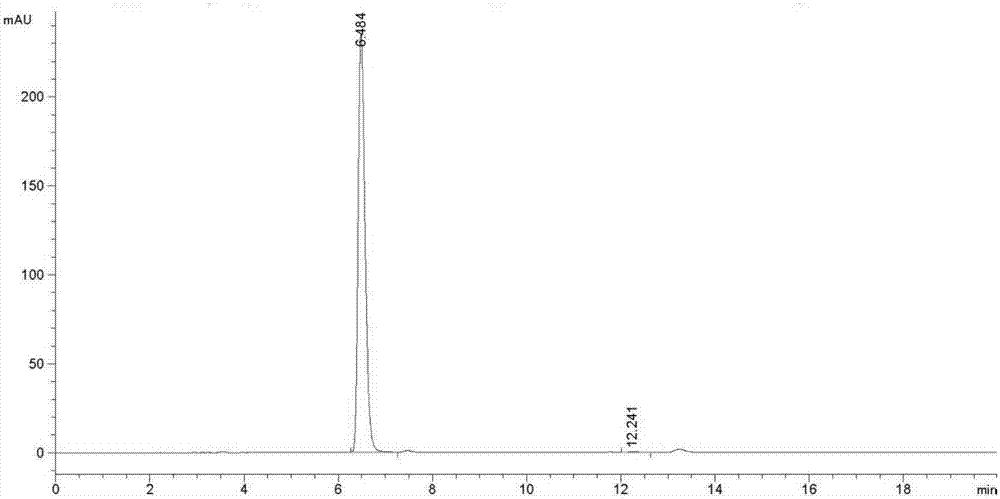
图1表示的是文献1方法得到的m3g(式i所示化合物)的hplc图(前者为m3g,后者为吗啡)。
图2表示的是本发明实施例1方法得到的m3g(式i所示化合物)的hplc图(前者为m3g,后者为吗啡)。
图3表示的是本发明实施例1制备的m3g(式i所示化合物)的1h-nmr图。
图4表示的是本发明实施例1制备的m3g(式i所示化合物)的质谱图。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。
在本发明的实施例中,1h-nmr检测仪器为brukerfourier400核磁共振波谱仪。
产品的纯度采用waters2695型高效液相色谱仪(hplc)进行分析,色谱条件为:分析柱:diamonsiltmc18(250mm×4.6mm);流动相:0.01mol·l-1磷酸二氢钾(0.5mmol·l-1的十二烷基磺酸钠)-乙腈(71:29);流速:0.65ml·min-1;紫外检测波长:210nm;进样量:10μl。
实施例1
(1)式ii所示化合物(r1为乙酰基)的合成
取10g吗啡加入乙腈中,加入7.1g三乙胺,降至0℃,滴加乙酸酐7.5g,控温10℃以下,滴毕,20~30℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为乙酰基)和式iv化合物(r5为三氟乙酰基)的合成
将第一步所得产物用150ml无水乙醇溶解,加入盐酸羟胺2.4g,20~30℃反应12小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用50ml乙腈溶解,控制反应温度-5~5℃,滴加5.9g三氟乙酸酐,滴加完毕,自然升温至室温,至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,浓减压缩,残留物用100ml新鲜的二氯甲烷溶解进行下一步反应。
(3)式vi所示化合物(r1为乙酰基,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的二氯甲烷溶液中加入式v所示的化合物(r2、r4为乙酰基,r3为甲基)17.2g,加入溴化锌5g,35~45℃反应24小时,过滤,反应液补加100ml二氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用异丙醇重结晶,0~5℃析晶12小时,过滤,得产品12.3g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的式vi所示化合物12.3g加入甲醇123ml,纯化水27ml,冷却至-50~-40℃,分批加入氢氧化钾4.8g,并在此温度下反应2小时,加入乙酸5.1g进行中和,升温至0~5℃析晶15小时,过滤,滤饼30℃减压干燥,得白色固体7.5g,使用50%的甲醇水溶液进行重结晶纯化,得m3g2.3g,hplc峰面积归一化法,测得其纯度为99.4%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例2
(1)式ii所示化合物(r1为苯甲酰基)的合成
取10g吗啡加入四氢呋喃中,加入2.8g氢化钠,降至0℃,滴加苯甲酰氯10.2g,控温10℃以下,滴毕,20~30℃反应1小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为苯甲酰基)和式iv化合物(r5为三氟甲磺酰基)的合成
将第一步所得产物用120ml无水甲醇溶解,加入苯肼3.8g,30~40℃反应10小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用100ml乙腈溶解,控制反应温度-10~0℃,滴加10g三氟甲磺酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml新鲜的二氯甲烷溶解进行下一步反应。
(3)式vi所示化合物(r1为苯甲酰基,r2为异丁酰基,r3为乙基)的合成
在步骤(2)所得的乙腈溶液中加入式v所示的化合物(r2为异丁酰基、r4为三氟乙酰基,r3为乙基)25.3g,加入三氟化硼乙醚10ml,40~60℃反应18小时,反应液补加200ml二氯甲烷,饱和碳酸氢钠溶液中和,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用异丙醇重结晶,0~5℃析晶12小时,过滤,得产品18.4g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的式vi所示化合物18.4g加入乙醇150ml,纯化水30ml,冷却至-40~-30℃,分批加入氢氧化钠4.5g,并在此温度下反应2小时,加入盐酸进行中和,升温至0~5℃析晶15小时,过滤,滤饼30℃减压干燥,得白色固体10.5g,进行柱层析纯化,洗脱剂为甲醇-水,梯度进行洗脱,收集洗脱液进行减压浓缩,用适量水重结晶,得m3g1.8g,hplc峰面积归一化法,测得其纯度为99.7%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例3
(1)式ii所示化合物(r1为3,5-二硝基苯甲酰基)的合成
取10g吗啡加入丙酮中,加入5.5g吡啶,降至0℃,滴加3,5-二硝基苯甲酰氯16.2g,控温10℃以下,滴毕,10~20℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为3,5-二硝基苯甲酰基)和式ⅵ化合物(r5为三氟乙酰基)的合成
将第一步所得产物用150ml异丙醇溶解,加入无水碳酸钾4.8g,50~60℃反应12小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用乙腈溶解,控制反应温度-5~5℃,滴加5.9g三氟乙酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml甲苯溶解进行下一步反应。
(3)式iv所示化合物(r1为三氟乙酰基,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的甲苯溶液中加入式v所示的化合物(r2为乙酰基,r4为溴,r3为甲基)18.2g,加入碳酸银15g,120~130℃反应2小时,过滤,滤液浓缩,加100ml二氯甲烷溶解,用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用丙酮重结晶,0~5℃析晶12小时,过滤,得产品12.8g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷12.8g加入异丙醇110ml,纯化水20ml,冷却至-30~-20℃,分批加入叔丁醇钾19.6g,并在此温度下反应1小时,加入乙酸5.1g进行中和,升温至0~5℃析晶12小时,过滤,滤饼30℃减压干燥,得白色固体6.5g,使用50%的乙腈水溶液进行柱层析纯化,得m3g0.8g,hplc峰面积归一化法,测得其纯度为99.6%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例4
(1)式ii所示化合物(r1为3-硝基苯甲酰基)的合成
取10g吗啡加入乙腈中,加入9.6g碳酸钾,降至0℃,滴加3-硝基苯甲酰氯13g,控温5℃以下,滴毕,10~20℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为硝基苯甲酰基)和式ⅵ化合物(r5为三氟乙酰基)的合成
将第一步所得产物用150ml无水甲醇溶解,加入硼酸钠13.5g,20~30℃反应1小时,过滤,滤液减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用50ml乙腈溶解,控制反应温度-5~5℃,滴加5.9g三氟乙酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml三氯甲烷溶解进行下一步反应。
(3)式iv所示化合物(r1为硝基苯甲酰基,r2为特戊酰基,r3为甲基)的合成
在步骤(2)所得的三氯甲烷溶液中加入式v所示的化合物(r2为特戊酰基、r4为三氟甲磺酰基,r3为甲基)17.5g,加入三甲基硅烷三氟甲磺酸酯三甲基硅烷三氟甲磺酸酯5ml,35~45℃反应36小时,过滤,反应液补加100ml三氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用异丙醇重结晶,0~5℃析晶12小时,过滤,得产品19.7g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷19.7g加入甲醇156ml,纯化水35ml,冷却至-20~-10℃,分批加入氢氧化钙6.4g,并在此温度下反应2.5小时,加入盐酸进行中和,升温至0~5℃析晶10小时,过滤,滤饼30℃减压干燥,得白色固体8.6g,使用甲醇、乙腈、水的混合溶液进行制备液相纯化,得m3g1.8g,hplc峰面积归一化法,测得其纯度为99.8%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例5
(1)式ii所示化合物(r1为异丁酰基)的合成
取10g吗啡加入乙腈中,加入10ml吗啉,降至0℃,滴加异丁酰氯13g,控温5℃以下,滴毕,10~20℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为异丁酰基)和式ⅵ化合物(r5为三氟甲磺酰基)的合成
将第一步所得产物用150ml乙二醇溶解,加入丁胺20g,50~60℃反应11小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用70ml乙腈溶解,控制反应温度-10~0℃,滴加10g三氟甲磺酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml苯溶解进行下一步反应。
(3)式iv所示化合物(r1为异丁酰基,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的甲溶液中加入式v所示的化合物(r2为乙酰基、r4为碘,r3为甲基)27.5g,加入三氯化铝7g,100~110℃反应5小时,过滤,滤液补加100ml三氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用丙酮重结晶,0~5℃析晶12小时,过滤,得产品19.7g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷19.7g加入异丙醇156ml,纯化水35ml,冷却至-10~0℃,分批加入氢氧化钡8.4g,并在此温度下反应2.5小时,加入盐酸进行中和,升温至0~5℃析晶10小时,过滤,滤饼30℃减压干燥,得白色固体7.4g,使用乙腈、水的混合溶液进行制备液相纯化,得m3g0.8g,hplc峰面积归一化法,测得其纯度为99.8%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例6
(1)式ii所示化合物(r1为对甲基苯磺酰基)的合成
取10g吗啡加入乙腈中,加入10ml三乙胺,降至0℃,滴加对甲基苯磺酰氯13.3g,控温5℃以下,滴毕,0~20℃反应6小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为对甲基苯磺酰基)和式ⅵ化合物(r5为三氟甲磺酰基)的合成
将第一步所得产物用150ml乙二醇溶解,加入硼氢化钠10g,40~60℃反应18小时,过滤,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用100ml乙腈溶解,控制反应温度-10~0℃,滴加10g三氟甲磺酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml二甲苯溶解进行下一步反应。
(3)式iv所示化合物(r1为三氟甲磺酰基,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的二甲苯溶液中加入式v所示的化合物(r2为乙酰基、r4为三氟甲磺酰基,r3为甲基)12.5g,加入溴化亚铜5g,140~160℃反应1小时,过滤,滤液补加100ml二氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用丙酮重结晶,0~5℃析晶12小时,过滤,得产品19.7g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷19.7g加入异丙醇156ml,纯化水35ml,冷却至-10~0℃,分批加入氢氧化钡8.4g,并在此温度下反应2.5小时,加入盐酸进行中和,升温至0~5℃析晶10小时,过滤,滤饼30℃减压干燥,得白色固体7.4g,使用乙腈、水的混合溶液进行制备液相纯化,得m3g0.8g,hplc峰面积归一化法,测得其纯度为99.8%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例7
(1)式ii所示化合物(r1为ch3o-co-co-)的合成
取10g吗啡加入四氢呋喃中,加入10g碳酸钠,降至0℃,滴加草酰氯单甲酯25g,控温5℃以下,滴毕,10~20℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为ch3o-co-co-)和式ⅵ化合物(r5为三氟甲磺酰基)的合成
将第一步所得产物用150ml乙醇溶解,持续通入氨气,10~20℃反应15小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用100ml乙腈溶解,控制反应温度-10~0℃,滴加10g三氟甲磺酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,残留物用100ml硝基甲烷溶解进行下一步反应。
(3)式iv所示化合物(r1为ch3o-co-co-,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的硝基甲烷溶液中加入式v所示的化合物(r2为乙酰基、r4为溴,r3为甲基)27.5g,加入溴化铜7g,100~110℃反应5小时,过滤,滤液补加100ml三氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用丙酮重结晶,0~5℃析晶12小时,过滤,得产品19.7g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷19.7g加入异丙醇156ml,纯化水35ml,冷却至--25~-10℃,分批加入氢氧化钡8.4g,并在此温度下反应2.5小时,加入盐酸进行中和,升温至0~5℃析晶10小时,过滤,滤饼30℃减压干燥,得白色固体7.4g,使用乙腈、水的混合溶液进行制备液相纯化,得m3g0.8g,hplc峰面积归一化法,测得其纯度为99.8%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
实施例8
(1)式ii所示化合物(r1为二氯乙酰基)的合成
取10g吗啡加入四氢呋喃中,加入10g碳酸钠,降至0℃,滴加二氯醋酸酐25g,控温5℃以下,滴毕,10~20℃反应5小时,将反应液倒入100ml二氯甲烷中,用饱和碳酸氢钠溶液中和,50ml纯化水、饱和氯化钠洗涤,无水硫酸镁干燥,过滤,减压浓缩。
(2)式iii所示化合物(r1为二氯乙酰基)和式ⅵ化合物(r5为三氟乙酰基)的合成
将第一步所得产物用150ml乙醇溶解,加入碳酸钠,60~70℃反应15小时,减压浓缩,浓缩物加入50ml水和120ml二氯甲烷,萃取分液,二氯甲烷层用饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,得浓缩物用乙腈溶解,控制反应温度-5~5℃,滴加5.9g三氟乙酸酐,滴加完毕,自然升温至室温至反应完毕,将反应体系直接倒入100ml二氯甲烷中,用碳酸氢钠溶液中和,二氯甲烷层用无水硫酸镁干燥,减压浓缩,浓缩物用100ml二氯甲烷和硝基甲烷的混合液溶解进行下一步反应。
(3)式iv所示化合物(r1为二氯乙酰基,r2为乙酰基,r3为甲基)的合成
在步骤(2)所得的硝基甲烷和二氯甲烷的混合溶液中加入式v所示的化合物(r2为乙酰基、r4为溴,r3为乙基)27.5g,加入溴化亚铜7g,100~110℃反应5小时,过滤,滤液补加100ml二氯甲烷,饱和氯化钠溶液洗涤,无水硫酸镁干燥,过滤,减压浓缩,浓缩物用丙酮重结晶,0~5℃析晶12小时,过滤,得产品19.7g。
(4)m3g(式i所示化合物)的合成
将步骤(3)所得的糖苷19.7g加入异丙醇156ml,纯化水35ml,冷却至--35~-20℃,分批加入氢氧化钡8.4g,并在此温度下反应2.5小时,加入盐酸进行中和,升温至0~5℃析晶10小时,过滤,滤饼30℃减压干燥,得白色固体7.4g,使用水进行重结晶相纯化,得m3g0.6g,hplc峰面积归一化法,测得其纯度为99.8%。
ms(lc-ms)462.1(m+h+)
1h-nmr(400mhz,dmso-d6),δ:2.32-2.44(m,3h,-ch,-ch2),3.16(s,3h,-nch3),4.71-4.73(d,1h,-ch),5.06-5.08(d,1h,-ch),5.21-5.24(m,1h,-ch),5.32(d,1h,-oh),5.55-5.57(d,1h,-ch),6.46-6.48(d,1h,-ch),6.70-6.72(d,1h,ch)。
- 还没有人留言评论。精彩留言会获得点赞!