基于微流控芯片和G‑四链体‑血红素DNA酶检测循环核酸用试剂盒及其制备方法和应用与流程
本发明属于微全分析系统技术领域,具体涉及基于微流控芯片和g-四链体-血红素dna酶检测循环核酸用试剂盒及其制备方法和应用。
背景技术:
生物大分子是构成生命的基础物质,包括蛋白质、核酸、碳氢化合物等。目前,生物大分子分析的检测技术主要有:(1)微阵列芯片技术(microarray);(2)静态微流控阵列芯片技术;(3)常规分子生物学技术。但是这几类技术普遍存在需要昂贵、精密的检测设备,检测时间长且普遍灵敏度不理想,不能实现微量样品的高通量检测,成本较高等缺点,制约了这些技术在生物大分子临床诊断领域获得更加广泛及深入的应用(j.mol.diagn.,2017,19:697-710;worldjgastrointestoncol.,2017,9:142-152;eur.rev.med.pharmacol.sci.,2016,20:2558-2564;methodsmol.biol.,2017,1634:283-303)。
动态微阵列是近年来发展的新兴研究领域,微流控微珠阵列芯片是动态微阵列研究领域中发展的一种新型芯片模式,它有机的将传统的微阵列芯片和微流控芯片结合起来,以功能化微球作为传感元件,以微流体作为试剂及样品传送方式实现微量检测(microchimicaacta,2015,182:661-669;biosensorsandbioelectronics,2014,57:22-29;biosensorsandbioelectronics,2013,42:23-30)。但是传统动态微阵列的开发仍然不能克服微量样品条件下高通量、高灵敏特性的难题。
技术实现要素:
有鉴于此,本发明的目的在于提供基于微流控芯片和g-四链体-血红素复合物的检测试剂盒及其制备方法和应用,所述检测试剂盒具有高通量、高检测灵敏度的特性。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了基于微流控芯片和g-四链体-血红素复合物检测循环核酸用试剂盒,包括以下组分:
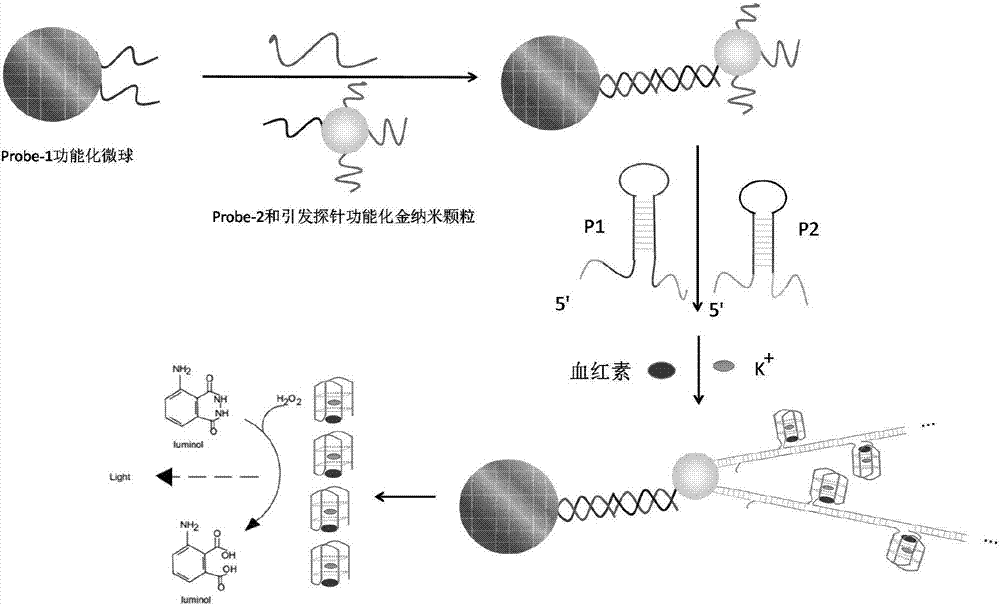
1)包被有在微型小室内的有功能化微球的微流控检测芯片;所述功能化微球为表面通过生物素-亲和素系统修饰有第一探针的微球;第一探针的5’端序列与循环核酸的5’端序列互补配对,所述第一探针的3’端序列修饰在微球上;
2)表面修饰有第二探针和引发探针的金纳米颗粒液;所述引发探针具有如序列表seqidno.1所示的核苷酸序列;第二探针的3’端序列与循环核酸的3’端序列互补配对,所述第二探针的5’端序列修饰在金纳米颗粒上;
3)第一发夹探针试剂;所述第一发夹探针具有如序列表seqidno.2所示的核苷酸序列;
4)第二发夹探针试剂;所述第二发夹探针试剂具有如序列表seqidno.3所示的核苷酸序列;
5)发光体系。
优选的,所述循环核酸为甲胎蛋白编码基因转录的mrna序列,所述第一探针具有如序列表seqidno.4所示的核苷酸序列,所述第二探针具有如序列表seqidno.5所示的核苷酸序列。
优选的,所述循环核酸为ceacam5编码基因转录的mrna序列,所述第一探针具有如序列表seqidno.6所示的核苷酸序列,所述第二探针具有如序列表seqidno.7所示的核苷酸序列。
优选的,所述循环核酸为ceacam7编码基因转录的mrna序列,所述第一探针具有如序列表seqidno.8所示的核苷酸序列,所述第二探针具有如序列表seqidno.9所示的核苷酸序列。
优选的,所述微流控检测芯片设置有微型小室,每个微型小室中包被有功能化微球的数量为10~25个。
优选的,所述微型小室呈阵列分布;所述微型小室为100~150μm宽、100~150μm长、30~50μm深。
优选的,每个功能化微球表面修饰有3×107条第一探针。
优选的,每个金纳米颗粒表面修饰有第二探针的数量为20~30条;每个金纳米颗粒表面修饰有引发探针的数量为200~300条。
优选的,所述第二探针与引发探针的摩尔比为1:8~12;所述金纳米颗粒液的浓度为23~24nmol/l。
本发明提供了所述试剂盒的制备方法,包括包被有功能化微球的微流控检测芯片的制备方法和表面修饰有第二探针和引发探针的金纳米颗粒的制备方法;
所述包被有功能化微球的微流控检测芯片的制备方法,包括以下步骤:
1)将亲和洗涤后的质量浓度为1%~3%的亲和素修饰微球液和0.3μmol/l生物素标记的第一探针溶液混合孵育,洗涤,得到功能化微球;
2)将所述步骤1)中功能化微球通过微球装载通道进入芯片中的微型小室中,固定,剥离微珠装载通道后,将试剂传送片基和微球固定阵列片基贴合,构建得到包被有功能化微球的微流控检测芯片;
所述表面修饰有第二探针和引发探针的金纳米颗粒液的制备方法,包括以下步骤:
a、将巯基修饰的第二探针溶液、巯基修饰的引发探针溶液、二硫苏糖醇溶液和金纳米颗粒混合,在4℃下静置16h;
b、向静置后的混合液中逐滴加入缓冲液,混匀静置,25~35℃放置24h后,得到表面修饰有第二探针和引发探针的金纳米颗粒液。
优选的,所述微球为聚苯乙烯;所述微球的粒径为15~25μm。
优选的,所述步骤1)中亲和素修饰微球液和生物素标记的第一探针溶液的体积比为42~45:2~4。
优选的,所述步骤1)中亲和素修饰微球中每个微球上修饰的亲和素的个数为0.8~1×107个。
优选的,所述步骤1)中亲和洗涤用亲和洗脱液包含以下含量组分:20mmol/l的tris,摩尔浓度为1mol/l的nacl,摩尔浓度为1mmol/l的edta,质量浓度为0.0005%tritonx-100;所述亲和洗脱液的ph值为7.5。
优选的,所述步骤a中巯基修饰的第二探针、巯基修饰的引发探针的摩尔比为1:9~11。
优选的,所述混合液与缓冲液的体积比为8~9:1。
优选的,所述缓冲液包括以下含量组分:摩尔浓度为2mol/lnacl,摩尔浓度为50mmol/ltris-hcl。
优选的,所述步骤b中静置后还包括固液分离、得到的沉淀物进行洗涤、沉淀和重悬,得到功能化的金纳米颗粒。
优选的,洗涤用溶液包括以下含量组分:摩尔浓度为10mm的tris-hcl,体积浓度为0.1%tween20,摩尔浓度为0.15mmol/l的nacl所述洗涤用溶液的ph值为7.4。
本发明提供了所述试剂盒或所述方法制备的检测试剂盒在检测循环核酸中的应用。
优选的,包括以下步骤:
1)将待测样品导入包被有功能化微球的微流控检测芯片中,洗脱,得到捕获有循环核酸的微流控检测芯片;
2)将表面修饰有第二探针和引发探针的金纳米颗粒液导入捕获有循环核酸微流控检测芯片中,洗脱,得到捕获有功能化金纳米颗粒的微流控检测芯片;
3)将第一发夹探针试剂和第二发夹探针试剂导入所述捕获有功能化金纳米颗粒的微流控检测芯片中,洗涤后,继续导入发光体系,孵育;
4)将得到的微流控检测芯片进行化学发光检测;根据检测结果和预定的标准曲线计算定量结果。
优选的,所述循环核酸包括甲胎蛋白、ceacam5或ceacam7编码基因转录的mrna序列。
优选的,甲胎蛋白编码基因转录的mrna序列具有如序列表seqidno.10所示的核苷酸序列。
优选的,ceacam5编码基因转录的mrna序列具有如序列表seqidno.11所示的核苷酸序列。
优选的,ceacam7编码基因转录的mrna序列具有如序列表seqidno.12所示的核苷酸序列。
本发明提供了基于微流控芯片和g-四链体-血红素复合物的检测循环核酸用试剂盒。以循环核酸为分析对象,以微流控动态微阵列技术为依托,以功能化金纳米颗粒表面杂交链式反应和g-四链体-血红素dna酶化学发光新体系为信号放大手段,发展以微观条件下功能化微球为传感元件的高灵敏生物大分子分析新技术。该试剂盒的建立能够较好的解决微量医学分析技术领域或微创、无创诊断领域中存在的微量样品条件下高通量、高灵敏特性难以并存的难题。该技术可实现疾病相关循环生物大分子的高灵敏检测、微创样品检测、可视化检测、不依赖复杂设备、成本低等特点,具有极大潜力发展为临床诊断技术。本发明提供的试剂盒,在循环核酸浓度在0.1pmol/l~500pmol/l之间时,呈良好的线性关系,回归方程为a=0.818c循环核酸+17.07,线性相关系数r2=0.997。以空白组的3倍标准偏差除以标准曲线的斜率得到本方法的检出限为0.05pmol/l。
附图说明
图1为本发明中微流控微珠阵列芯片的结构示意图及微结构显微照片;图1-a为芯片的结构示意图;图1-b为微型小室为单位构成的阵列;图1-c为功能化微球通过微珠装载片基;图1-d为微球从宽缝流入后不能从另一端的窄缝流出,聚集在小室中;图1-e为微球固定阵列片基和试剂传送片基贴合构建的检测芯片;
图2为基于微流控芯片和杂交链式反应的g-四链体-血红素dna酶与金纳米颗粒协同催化化学发光新体系检测循环核酸原理图;
图3为实施例2中协同催化化学发光新体系检测甲胎蛋白编码基因的mrna分子的标准曲线;
图4为实施例3中协同催化化学发光新体系检测甲胎蛋白循环mrna血液样品的结果;
图5为实施例5中协同催化化学发光新体系检测癌胚抗原5编码基因的mrna分子的标准曲线;
图6为实施例7中协同催化化学发光新体系检测癌胚抗原5循环mrna血液样品的结果
图7为实施例8中协同催化化学发光新体系检测癌胚抗原7编码基因的mrna分子的标准曲线。
图8为实施例9中协同催化化学发光新体系检测癌胚抗原7循环mrna血液样品的结果。
具体实施方式
本发明提供了基于微流控芯片和g-四链体-血红素复合物的检测循环核酸用试剂盒,包括以下组成:
1)包被在微型小室中的功能化微球的微流控检测芯片;所述功能化微球为表面通过生物素-亲和素系统修饰有第一探针的微球;第一探针的5’端序列与循环核酸的5’端序列互补配对,所述第一探针的3’端序列修饰在微球上;
2)表面修饰有第二探针和引发探针的金纳米颗粒液;所述引发探针具有如序列表seqidno.1所示的核苷酸序列;第二探针的3’端序列与循环核酸的3’端序列互补配对,所述第二探针的5’端序列修饰在金纳米颗粒上;
3)第一发夹探针试剂;所述第一发夹探针具有如序列表seqidno.2所示的核苷酸序列;
4)第二发夹探针试剂;第二发夹探针试剂具有如序列表seqidno.3所示的核苷酸序列;
5)发光体系。
本发明提供的检测循环核酸用试剂盒包括包被有功能化微球的微流控检测芯片。所述微流控检测芯片设置有若干个微型小室(如图1)。所述微型小室优选呈阵列分布。所述微型小室优选为100~150μm宽、100~150μm长、30~50μm深,更优选为120μm宽、120μm长、40μm深。所述微流控检测芯片优选还设置有加样池和废液池。加样池和废液池优选为pdms材料。所述加样池和废液池的规格独立优选为:1~3mm宽、1~3mm长、1~2mm深。所述加样池和废液池分布在微型小室两端,并且用试剂传送片基将加样池和废液池分别与微型小室相连。所述试剂传送片基使用前优选进行封闭。所述封闭用溶液为bsa溶液。试剂传送片基为pdms片基包含的微结构阵列覆盖通道的尺寸为130μm宽、130μm长、10μm深。
本发明中,每个微型小室中包被有功能化微球的数量优选为10~25个,更优选为20个。所述微型小室的材料优选为聚二甲基硅氧烷。本发明中,预先将功能化微球利用微球装载通道固定在30微米深微室阵列中,再与试剂传送片基上的10微米深的覆盖区匹配,由于微球直径为15微米,微球只能限制在小室中,不能流入试剂传送片基上的10微米深的试剂通道,从而实现固定了微球。
本发明中,所述微球优选为聚苯乙烯微球。所述微球的粒径优选为15~25条,更优选为15μm。
本发明中,每个功能化微球表面修饰的第一探针的数量优选2.8~3.5×107条,更优选为3×107条。
本发明中,所述功能化微球为表面通过生物素-亲和素系统修饰有第一探针的微球。本发明中,所述功能化微球由亲和素修饰的微球和生物素修饰的第一探针按照摩尔比为1:4的比例混合得到。本发明中,所述亲和素修饰的微球购买自bangslab公司。所述生物素修饰的第一探针委托上海生工生物工程有限公司合成。
本发明提供的检测循环核酸用试剂盒包括表面修饰有第二探针和引发探针的金纳米颗粒液。本发明中,每个金纳米颗粒表面修饰有第二探针的数量优选为20~30条,更优选为25条;每个金纳米颗粒表面修饰有引发探针的数量为200~300条,更优选为250条。本发明中,所述第二探针与引发探针的摩尔比优选为1:8~12,更优选为1:10。所述第二探针与引发探针均进行巯基修饰。所述第二探针和引发探针上的巯基与金纳米颗粒表面通过配位键充分结合。
本发明中,当所述循环核酸为甲胎蛋白编码基因转录的mrna序列时,所述第一探针具有如序列表seqidno.4所示的核苷酸序列,所述第二探针具有如序列表seqidno.5所示的核苷酸序列。
本发明中,当所述循环核酸为ceacam5编码基因转录的mrna序列时,所述第一探针具有如序列表seqidno.6所示的核苷酸序列,所述第二探针具有如序列表seqidno.7所示的核苷酸序列。
本发明中,当所述循环核酸为ceacam7编码基因转录的mrna序列时,所述第一探针具有如序列表seqidno.8所示的核苷酸序列,所述第二探针具有如序列表seqidno.9所示的核苷酸序列。
本发明提供的检测循环核酸用试剂盒包括第一发夹探针试剂和第二发夹探针试剂。所述第一发夹探针具有如序列表seqidno.2所示的核苷酸序列。第二发夹探针试剂具有如序列表seqidno.3所示的核苷酸序列;第一发夹探针试剂和第二发夹探针试剂优选进行人工合成。本发明中,所述第一发夹探针试剂和第二发夹探针试剂委托上海生工生物工程有限公司进行合成。第一发夹探针试剂的浓度优选为0.4~0.6umol/l,更优选为0.5umol/l。第二发夹探针试剂的浓度优选为0.4~0.6umol/l,更优选为0.5umol/l。第一发夹探针试剂和第二发夹探针试剂通过引发探针启动杂交链式反应,在金纳米颗粒表面形成带有大量分子间裂分式g-四链体序列的dna纳米线,以便后期加入血红素后,组装成g-四链体-血红素dna酶。
本发明提供的检测循环核酸用试剂盒包括发光体系。所述发光体系为氯化血红素、鲁米诺和双氧水的化学发光混合体系。所述混合体系中鲁米诺的浓度为0.4~0.6mmol/l,更优选为0.5mmol/l。所述混合体系中双氧水的浓度优选为25~35mmol/l,更优选为30mmol/l。所述混合体系中氯化血红素的浓度优选为70~80nmol/l,更优选为75nmol/l。
本发明提供了所述试剂盒的制备方法,包括包被有功能化微球的微流控检测芯片的制备方法和表面修饰有第二探针和引发探针的金纳米颗粒的制备方法。
本发明中所述包被有功能化微球的微流控检测芯片的制备方法,包括以下步骤:
1)将亲和洗涤后的质量浓度为1%~3%的亲和素修饰微球液和0.3μmol/l生物素标记的第一探针溶液混合孵育,洗涤,得到功能化微球;
2)将所述步骤1)中功能化微球通过微球装载通道进入芯片中的微型小室中,固定,剥离微珠装载通道后,将试剂传送片基和微球固定阵列片基贴合,构建得到包被有功能化微球的微流控检测芯片。
本发明将亲和洗涤后的质量浓度为1%~3%的亲和素修饰微球液和0.3μmol/l生物素标记的第一探针溶液混合孵育,洗涤,得到功能化微球。
本发明对亲和洗涤的方式没有特殊限制,采用本领域技术人员所熟知的洗涤方式即可。亲和洗涤用溶液为亲和洗脱液。所述亲和素修饰微球液与洗涤液的体积比为1:1。所述亲和洗脱液包含以下含量组分:20mmol/l的tris,摩尔浓度为1mol/l的nacl,摩尔浓度为1mmol/l的edta,质量浓度为0.0005%tritonx-100;所述亲和洗脱液的ph值为7.5。洗涤后,本发明通过离心方法去除洗涤液。所述离心的转速优选为3000~4000rpm,更优选为3500rpm。所述离心的时间优选为3~8min,更优选为5min。收集离心后的沉淀,用亲和洗脱液重悬。所述洗涤的次数优选为2~3次。
本发明对所述混合孵育的方法没有特殊限制,采用本领域技术人员所熟知的混合孵育方案即可。本发明中,所述混合孵育的温度优选为23~27℃,更优选为25℃。所述混合孵育的时间优选为10~15min,更优选为12min。所述亲和素修饰微球液和生物素标记的第二探针溶液的体积比为42~45μl:2~4μl。
本发明对洗涤的方式没有特殊限制,采用本领域技术人员所熟知的洗涤方式即可。所述洗涤能够去除未结合在微球上的生物素标记的第一探针分子。洗涤后,将功能化微球悬浮于100μl亲和洗脱液。
本发明中,所述亲和素修饰微球中每个微球上修饰的亲和素的个数为0.8~1×107个。生物素标记的第一探针分子中,每个第一探针由一个生物素标记。
得到功能化微球后,本发明将所述功能化微球通过微球装载通道进入芯片中的微型小室中,固定,剥离微球装载通道后,将试剂传送片基和微球固定阵列片基贴合,构建得到包被有功能化微球的微流控检测芯片。
本发明中,不连通的微球装载通道与微型小室相对贴合,在微球装载通道两侧形成2个缝隙,微球从宽的缝隙流入小室,但不能从小的缝隙流出,将微球留在微型小室中。所述微球装载通道优选进行封闭。所述封闭用溶液为bsa溶液。所述bsa溶液的质量浓度优选为1%~3%,更优选为2%。所述微球装载通道优选聚二甲基硅氧烷片基(pdms)。
本发明中,所述表面修饰有第二探针和引发探针的金纳米颗粒液的制备方法,包括以下步骤:
a、在巯基修饰的第二探针溶液、巯基修饰的引发探针溶液、二硫苏糖醇溶液和金纳米颗粒混合,在4℃下静置16h;
b、向静置后的混合液逐滴加入缓冲液,混匀静置,22~27℃放置24h后,得到表面修饰有第二探针和引发探针的金纳米颗粒液。
本发明对所述混合的方案没有特殊限制,采用本领域技术人员所熟知的混合方案即可。本发明中,所述巯基修饰的第二探针、巯基修饰的引发探针的摩尔比优选为1:10。所述巯基修饰的第二探针和巯基修饰的引发探针溶液委托上海生工生物工程有限公司合成。二硫苏糖醇溶液的添加体积为17.6ul。所述二硫苏糖醇溶液的摩尔浓度优选为1mmol/l。
混合后,本发明向静置后的混合液中逐滴加入缓冲液,混匀静置,25~35℃放置24h后,得到表面修饰有第二探针和引发探针的金纳米颗粒液。
本发明将得到的混合液与缓冲液的体积比优选为8~9:1。所述缓冲液优选包括以下含量组分:摩尔浓度为2mol/lnacl,摩尔浓度为50mmol/ltris-hcl。
本发明中,所述静置后优选还包括固液分离、得到的沉淀物进行洗涤、沉淀和重悬。洗涤用溶液优选包括以下含量组分:摩尔浓度为10mm的tris-hcl,体积浓度为0.1%tween20,摩尔浓度为0.15mmol/l的nacl所述洗涤用溶液的ph值为7.4。
本发明提供了所述试剂盒或所述方法制备的试剂盒在检测循环核酸中的应用。
本发明中,优选包括以下步骤:
1)将待测样品导入包被有功能化微球的微流控检测芯片中,洗脱,得到捕获有循环核酸的微流控检测芯片;
2)将表面修饰有第二探针和引发探针的金纳米颗粒液导入捕获有循环核酸的微流控检测芯片中,洗脱,得到捕获有功能化金纳米颗粒的微流控检测芯片;
3)将第一发夹探针试剂和第二发夹探针试剂导入所述捕获有功能化金纳米颗粒的微流控检测芯片中,洗涤后,继续导入化学发光体系,孵育;
4)微流控检测芯片进行化学发光检测。
本发明将待测样品导入包被有功能化微球的微流控检测芯片中,洗脱,得到捕获有循环核酸的微流控检测芯片。
本发明中,导入待测样品的体积优选为10~20μl,更优选为15μl。导入待检测样品后,优选反应1~1.5h。所述反应的温度优选为35~38℃,更优选为37℃。
本发明对洗涤的方法没有特殊限制,采用本领域技术人员所熟知的洗涤方法即可。所述洗脱用溶液为0.8~1.2%的bsa的洗涤液。所述洗涤时间优选为4~8min,更优选为5min。所述洗涤液的体积优选为15~25μl。
导入待检测样品后,本发明将表面修饰有第二探针和引发探针的金纳米颗粒液导入洗脱后的微流控检测芯片中,洗脱,得到捕获有功能化金纳米颗粒的微流控检测芯片。
本发明中,所述导入的表面修饰有第二探针和引发探针的金纳米颗粒液的体积优选为4~8μl,更优选为5μl。导入上述液体后,优选反应1~1.5h。所述反应的温度优选为35~38℃,更优选为37℃。所述洗脱用溶液为hepes缓冲液。所述hepes缓冲液的摩尔浓度优选为10nmol/l。所述洗涤时间优选为4~8min,更优选为5min。所述洗涤液的体积优选为15~25μl。
得到捕获有功能化金纳米颗粒的微流控检测芯片后,本发明将第一发夹探针试剂和第二发夹探针试剂导入所述捕获有功能化金纳米颗粒的微流控检测芯片中,洗涤后,继续导入发光体系,孵育。
本发明中,导入的第一发夹探针试剂和第二发夹探针试剂的体积优选为8~12ul,更优选为10ul。第一发夹探针试剂和第二发夹探针试剂的浓度独立为0.5μmol/l。
本发明中,所述导入第一发夹探针和第二发夹探针试剂后,反应温度优选为25-30℃。所述反应的时间优选为5~7h,更优选为6h。
本发明中,洗涤方案跟上述洗涤方案相同。所述洗涤是洗去未结合第一发夹探针和第二发夹探针。
本发明中,所述导入发光体系的体积优选为18~23μl,更优选为20μl。
本发明中,所述孵育的温度优选为25~35℃,更优选为30℃。
微流控检测芯片孵育后,本发明将孵育后的微流控检测芯片进行化学发光检测。
本发明中,所述化学发光检测的波长优选为425nm。根据上述检测方法得到标准曲线。标样来源为上述循环核酸的标准品。
本发明中,化学发光强度根据式i计算。所述式i为δa=a-a0,其中a为样品测量值,a0为循环核酸浓度为0时的背景值。
本发明中,所述检测样品中循环核酸浓度在0.1pmol/l~500pmol/l之间时,呈良好的线性关系,回归方程为δa=0.818c循环核酸+17.07,线性相关系数r2=0.997。以空白组的3倍标准偏差除以标准曲线的斜率得到本方法的检出限为0.05pmol/l。
下面结合实施例对本发明提供的基于微流控芯片和g-四链体-血红素dna酶检测循环核酸用试剂盒及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
制备甲胎蛋白编码基因的mrna检测用试剂盒的方法
采用15μm亲和素修饰聚苯乙烯微球作为第一探针固定的固相界面,取100μl浓度为2%的亲和素修饰微球于离心管中,用100μl亲和洗脱液(20mmtrisph值7.5,1mnacl,1mmedta,0.0005%tritonx-100)洗涤两次,离心条件为3500rpm,5min,去上清;分别加入44μl的亲和洗脱液、3μl0.3μm生物素修饰的第一探针(序列见表1),常温孵育10~15min;通过离心洗涤方法去除未结合的分子并将功能化微球悬浮于100μl亲和洗脱液。第一探针通过修饰的生物素与微球表面的亲和素特异性结合并固定在微球表面,从而形成了具有目标分子检测能力的功能化聚苯乙烯微球。每个微球表面修饰3×107个生物素化的分子。
将设计的芯片结构图样通过绘图软件(coreldraw9.0)绘制出来,并以2400dpi的分辨率打印在kodak的菲林胶片上制备出芯片的光掩膜;然后将光掩膜的图案通过紫外曝光的方法转移到覆盖有光刻胶的pcb板上,并用化学刻蚀方法在曝光pcb板上制备出芯片的阳模板;最后将聚二甲基硅氧烷前聚体与固化剂(与聚二甲基硅氧烷前聚体是配套的)按10:1比例混合后于真空泵中除去气泡,然后平铺在芯片阳模板上(厚约1mm)。置于65℃烘箱中3h,待固化后取出,将聚二甲基硅氧烷(pdms)片基从阳模板上剥离下来。芯片的结构示意图如图1-a所示,功能化微球通过微珠装载片基(图1-c)(微球装载片基也是pdms的,带有不连通的微球装载通道,该片基中的通道壁使用bsa封闭,微球不会与通道壁结合,流入微型小室中),固定(不连通的微球装载通道与微型小室相对贴合,在微球装载通道两侧形成2个缝隙,微球从宽的缝隙流入小室,但不能从小的缝隙流出,将微球留在小室中)在以微型小室(图1-b)为单位构成的阵列中,微球从宽缝流入后不能从另一端的窄缝流出(图1-d),聚集在小室中;功能化微珠固定后,剥离微珠装载片基并将微球固定阵列片基(微球固定阵列片基包含有小室阵列,不同小室中装入不同功能化微球后形成阵列,微球会留在小室中)和试剂传送片基贴合构建检测芯片(图1-e)。设计的微流控芯片中,微珠固定微结构阵列由多个单独的小室构成,单个小室的尺寸为100μm宽、100μm长、30μm深;试剂传送片基为pdms片基包含的微结构阵列覆盖通道的尺寸为130μm宽、130μm长、10μm深。
金纳米颗粒功能化
在一支离心管中加入4ul巯基修饰(巯基与au纳米颗粒形成稳定的s-au半共价键,这是自发形成的,不需要特殊控制,这也是au表面自组装的常规技术)的第二探针和40ul巯基修饰的引发探针(1:10)(序列见表1),17.6ul1mm的dtt(二硫苏糖醇),603ul浓度为23.58nm的金纳米颗粒,4℃下混匀静置16h,使核酸适配体和引发探针上的巯基与金纳米颗粒表面通过配位键充分结合。然后逐渐向上述溶液中滴加80ul缓冲液a(2mnacl,50mmtris-hcl)混匀静置,室温放置24h后,将功能化金纳米颗粒溶液在17000转下离心分离35min,将金纳米粒子与未结合的核酸适配体分离,去除上层清液,用缓冲液b(10mmtris-hcl,0.1%tween20,0.15mmol/l的nacl,ph=7.4)洗涤沉淀,将功能化金纳米颗粒分散到1ml缓冲溶液b中,保存于4℃暗处,备用。
表1甲胎蛋白编码基因的mrna检测用核酸探针一览表
实施例2
检测甲胎蛋白编码基因的mrna分子的标准曲线的制备方法
将浓度为0.1pm~10nm甲胎蛋白序列(序列表中seqidno.10)加入缓冲体系中构建15μl杂交溶液,包括:10mmtris-hcl(ph7.5),750mmnacl,0.025%tween20,在压力驱动下通过包含功能化微珠阵列的微流控微珠阵列芯片检测区,常温下孵育30min,用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;流入15μl含有2nm第二探针上和引发探针修饰的金纳米颗粒,常温下孵育30min。用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;将0.5um第一发夹探针和第二发夹探针的混合溶液10ul流入反应井中,孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nm氯化血红素,0.5mm鲁米诺,30mmh2o2混合溶液)流入检测区进行发光检测,最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
定义δa=a-a0,其中a为样品测量值,a0为循环核酸浓度为0时的背景值,当循环核酸浓度在0.1pm~50pm之间时,呈良好的线性关系(图3),回归方程为δa=0.818c循环核酸+17.07,线性相关系数r2=0.997。以空白组的3倍标准偏差除以标准曲线的斜率得到本方法的检出限为0.05pm。
实施例3
采集5ml新鲜血样,加入10ml含有3%的右旋糖苷和0.9%nacl的溶液轻轻摇动并在室温孵育20min。12000rpm离心后去上清,得到有核细胞。采用pbs溶液洗涤后,采用常规的trizol试剂提取总rna,最后总rna溶解在15μldepc水中待测。
取15ul上述不同浓度甲胎蛋白编码基因的mrna样品流入微室内,在37℃下反应1h。用含1%的bsa的洗涤液冲洗5min,再加入5ul的1.5nm浓度的功能化的金纳米颗粒试剂,在37℃下反应1h,用1%的bsa的洗涤液冲洗5分钟。分别将0.5um发夹1和0.5um发夹2的混合溶液10ul流入微型小室中,30℃孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nmhemin,0.5mmluminol,30mmh2o2)流入检测区进行发光检测(425nm),最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
3个血液样本中循环甲胎蛋白mrna检测结果如图4所示,根据建立的标准曲线,其浓度分别为:样本1甲胎蛋白mrna浓度为10pmol/l,样本2甲胎蛋白mrna浓度为21.5pmol/l,样本3甲胎蛋白mrna浓度为13.2pmol/l。
对比例1
甲胎蛋白mrna常规检测方法主要有荧光定量rcr法、等温扩增法等,选择具有代表性的研究成果与本发明进行比对,如表2所示,荧光定量rcr法和等温扩增法由于对甲胎蛋白mrna进行指数扩增,灵敏度高于本发明,但是通过引物进行扩增,存在重复性差、开放检测体系导致的假阳性率高的问题,在实际应用中存在较大的局限;本发明直接对甲胎蛋白mrna进行捕获,随后进行信号放大,尤其是在封闭的微流控芯片内进行反应,结果可重复性高、假阳性率低,另外采用化学发光体系,不需要依赖大型检测设备,容易发展为现场检测技术。
表2甲胎蛋白mrna常规检测方法与本发明的比较结果
实施例4
按照实施例1所述方法制备检测用试剂盒,功能化微球上修饰的探针如表2中第一探针所示,纳米金颗粒上修饰的第二探针如表3中第二探针所示,制备得到检测癌胚抗原5(ceacam5)编码基因的mrna检测用试剂盒。
表3癌胚抗原5编码基因的mrna检测用核酸探针一览表
实施例5
检测癌胚抗原5(ceacam5)编码基因的mrna分子的标准曲线的制备方法
将含有浓度为0.1pm~10nm癌胚抗原5序列(序列表中seqidno.11)加入缓冲体系中构建15μl杂交溶液,包括:10mmtris-hcl(ph7.5),750mmnacl,0.025%tween20,在压力驱动下通过包含功能化微珠阵列的微流控微珠阵列芯片检测区,常温下孵育30min,用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;流入15μl含有2nm第二探针和引发探针修饰的金纳米颗粒,常温下孵育30min。用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;将0.5um第一发夹探针和第二发夹探针的混合溶液10ul流入反应井中,孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nm氯化血红素,0.5mm鲁米诺,30mmh2o2混合溶液)流入检测区进行发光检测,最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
定义δa=a-a0,其中a为样品测量值,a0为循环核酸浓度为0时的背景值,当循环核酸浓度在0.1pm~50pm之间时,呈良好的线性关系(图5),回归方程为δa=0.858c循环核酸+16.00,线性相关系数r2=0.992。以空白组的3倍标准偏差除以标准曲线的斜率得到本方法的检出限为0.03pm。
实施例6
采集5ml新鲜血样,加入10ml含有3%的右旋糖苷和0.9%nacl的溶液轻轻摇动并在室温孵育20min。12000rpm离心后去上清,得到有核细胞。采用pbs溶液洗涤后,采用常规的trizol试剂提取总rna,最后总rna溶解在15μldepc水中待测。
取15ul上述不同浓度ceacam5编码基因的mrna样品流入微室内,在37℃下反应1h。用含1%的bsa的洗涤液冲洗5min,再加入5ul的1.5nm浓度的功能化的金纳米颗粒试剂,在37℃下反应1h,用1%的bsa的洗涤液冲洗5分钟。分别将0.5um发夹1和0.5um发夹2的混合溶液10ul流入微型小室中,30℃孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nmhemin,0.5mmluminol,30mmh2o2)流入检测区进行发光检测(425nm),最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
3个血液样本中循环癌胚抗原5mrna检测结果如图6所示,根据建立的标准曲线,其浓度分别为:样本1癌胚抗原5mrna浓度为26.29pmol/l,样本2癌胚抗原5mrna浓度为0.27pmol/l,样本3癌胚抗原5mrna浓度为9.52pmol/l。
实施例7
按照实施例1所述方法制备检测用试剂盒,功能化微球上修饰的探针如表2中第一探针所示,纳米金颗粒上修饰的第二探针如表4中第二探针所示,制备得到检测癌胚抗原7(ceacam7)编码基因的mrna检测用试剂盒。
表4癌胚抗原7编码基因的mrna检测用核酸探针一览表
实施例8
检测癌胚抗原7(ceacam7)编码基因的mrna分子的标准曲线的制备方法
将含有浓度为0.1pm~10nm癌胚抗原7序列(序列表中seqidno.12)加入缓冲体系中构建15μl杂交溶液,包括:10mmtris-hcl(ph7.5),750mmnacl,0.025%tween20,在压力驱动下通过包含功能化微珠阵列的微流控微珠阵列芯片检测区,常温下孵育30min,用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;流入15μl含有2nm第二探针上和引发探针修饰的金纳米颗粒,常温下孵育30min。用55℃te缓冲液(10mmtris–hcl,ph8.0,1mmedta)洗涤5min;将0.5um第一发夹探针和第二发夹探针的混合溶液10ul流入反应井中,孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nm氯化血红素,0.5mm鲁米诺,30mmh2o2混合溶液)流入检测区进行发光检测,最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
定义δa=a-a0,其中a为样品测量值,a0为循环核酸浓度为0时的背景值,当循环核酸浓度在0.1pm~50pm之间时,呈良好的线性关系(图7),回归方程为δa=0.935c循环核酸+16.04,线性相关系数r2=0.989。以空白组的3倍标准偏差除以标准曲线的斜率得到本方法的检出限为0.04pm。
实施例9
采集5ml新鲜血样,加入10ml含有3%的右旋糖苷和0.9%nacl的溶液轻轻摇动并在室温孵育20min。12000rpm离心后去上清,得到有核细胞。采用pbs溶液洗涤后,采用常规的trizol试剂提取总rna,最后总rna溶解在15μldepc水中待测。
取15ul上述不同浓度ceacam7编码基因的mrna样品流入微室内,在37℃下反应1h。用含1%的bsa的洗涤液冲洗5min,再加入5ul的1.5nm浓度的功能化的金纳米颗粒试剂,在37℃下反应1h,用1%的bsa的洗涤液冲洗5分钟。分别将0.5um发夹1和0.5um发夹2的混合溶液10ul流入微型小室中,30℃孵育6个小时。10nmhepes缓冲液冲洗5min,将未反应的发夹1和发夹2除去。取20ul发光体系(75nmhemin,0.5mmluminol,30mmh2o2)流入检测区进行发光检测(425nm),最后利用荧光显微镜ccd拍摄检测区由于催化产生的蓝光并进行定量。
3个血液样本中循环癌胚抗原7mrna检测结果如图8所示,根据建立的标准曲线,其浓度分别为:样本1癌胚抗原7mrna浓度为0.59pmol/l,样本2癌胚抗原7mrna浓度为0.56pmol/l,样本3癌胚抗原7mrna浓度为1.25pmol/l。
对比例2
癌胚抗原mrna常规检测方法主要有荧光定量rcr法、巢式pcr等,选择具有代表性的研究成果与本发明进行比对,如表5所示,荧光定量rcr法和巢式pcr由于对癌胚抗原5mrna进行指数扩增,灵敏度高于本发明,但是通过引物进行扩增,存在重复性差、开放检测体系导致的假阳性率高的问题,在实际应用中存在较大的局限;本发明直接对癌胚抗原mrna进行捕获,随后进行信号放大,尤其是在封闭的微流控芯片内进行反应,结果可重复性高、假阳性率低,另外采用化学发光体系,不需要依赖大型检测设备,容易发展为现场检测技术。
表5癌胚抗原mrna常规检测方法与本发明的比较结果
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>湖南工程学院
<120>基于微流控芯片和g-四链体-血红素dna酶检测循环核酸用试剂盒及其制备方法和应用
<160>12
<170>siposequencelisting1.0
<210>1
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>1
ttttatttatttagaagaaggtgtttaagta31
<210>2
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>2
agggcgggtgggtgtttaagttggagaattgtacttaaacaccttcttcttgggt55
<210>3
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>3
tgggtcaattctccaacttaaactagaagaaggtgtttaagttgggtagggcggg55
<210>4
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>4
tctgttatttgtggcttttgcttcacccccccccctttttttttt45
<210>5
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttttttttttccccccccccctcgttttgtcttctcttcccctga45
<210>6
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>6
agactgtgattgtcttgactgtagtcccccccccctttttttttt45
<210>7
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>7
ttttttttttcccccccccccatccttgtcctccacgggttt42
<210>8
<211>48
<212>dna
<213>人工序列(artificialsequence)
<400>8
attttggacttccatcattcataggttacccccccccctttttttttt48
<210>9
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>9
ttttttttttccccccccccattaacactactgtcagttaccatc45
<210>10
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>10
tgaagcaaaagccacaaataacagatcaggggaagagaagacaaaacgag50
<210>11
<211>47
<212>dna
<213>人工序列(artificialsequence)
<400>11
actacagtcaagacaatcacagtctaaacccgtggaggacaaggatg47
<210>12
<211>53
<212>dna
<213>人工序列(artificialsequence)
<400>12
taacctatgaatgatggaagtccaaaatgatggtaactgacagtagtgttaat53
- 还没有人留言评论。精彩留言会获得点赞!