一种评估预后和预测恶性疾病患者对免疫治疗的反应的方法与流程
本专利申请要求德国专利申请第de102016005947.8号的优先权,其公开内容通过引用并入本文。
序列表
本申请包括作为说明书的一部分的根据wipost.25标准的txt格式的具有485个序列的电子序列表,其内容通过引用并入本文。
本发明涉及肿瘤学领域的分子诊断方法,利用该方法可以评估患有恶性疾病的患者的预后,或者可以预测患有恶性疾病的患者对免疫治疗的反应。本发明还涉及分子诊断方法,其允许个性化选择合适的用于这些患者的免疫治疗的药物。因此,本发明还涉及将这些方法用于预后确定、预测或个性化药物选择,以及用于个性化选择药物的预后和/或预测生物标志物或生物标记物。此外,本发明还涉及用于实施所述方法或用于所述用途的试剂盒。特别地,本发明限于体外的方法和应用。
背景技术:
为患者选择正确的治疗方法是现代医学的核心问题。一方面,选择正确的治疗需要最精确地估计患者在不依赖任何可能的治疗下最可能的疾病病程。在该预后的基础上,临床医生可以决定,例如,是否在可能有利的疗程的情况下进行相对保守的治疗,或者在可能不利的疗程的情况下进行相对激进的治疗。因此,对疾病进程的可靠评估将能够优化治疗。特别是,患者可以从更有针对性的治疗中受益。
评估患有恶性疾病(癌症)的患者的预后在目前是不太可能的。通常,基于恶性疾病的进展速度估计患者的预后。为此考虑例如转移的存在、肿瘤大小、和浸润进入淋巴和血管的参数。这些肿瘤阶段评估程序对于每个肿瘤实体是不同的。对于少数肿瘤,还有其他预后因素,例如依据gleason确定在前列腺癌患者中的生长模式。总之,这种对恶性疾病患者预后的确定不够可靠且难以标准化。
预后生物标志物有可能解决这个问题。us2014/0011702a1公开了基因dgki,mgmt和sdpr的甲基化分析,可以用来进行胶质母细胞瘤患者的预测和预后。us2006/0121467a1描述了一种方法,其包括基因stmn1、sfn、s100a2、tgfbr2、tp53、ptgs2、fgfr1、syk、pitx2、grin2d、psa、cga、cyp2d6、msmb、cox7a2l、vtn、prkcd、onecut2、wbp11、dag1、erbb2、tff1、tmeff2、esr1、rassf1、pitx2、psat1和pcaf的甲基化,用于预测乳腺癌激素治疗的治疗反应。从us2015/0051084a1中可知基因hspb1的甲基化用于确定前列腺癌患者的预后。在wo2011/051414a1中是利用基因clk3、mtmr4、nfe2l1、perld1、akap2、anapc1、ankrd47、atf7、atp5g1、bcl2、c9orf3、coil、csf2、dmp1、e2f3、elmo2、erbb2、gabrg2、gpc5、hspa6、kiss1、march3、kiaa0100、mro、msx1、ncoa6、papola、pdgfra、rpl23a、rufy3、sox3、tp53、tspyl1、ubxd3和znf420的甲基化,用于预后和预测卵巢癌患者对铂治疗的反应。
另一方面,可靠预测患者对免疫疗法的反应具有很大的临床和经济效益。当前肿瘤学的一项突破是所谓的免疫检查点抑制剂的免疫疗法,其即使在晚期肿瘤疾病中也显示出优异的结果。然而,只有相对较小比例的患者对这些疗法有反应。预测生物标志物可以预测对这些疗法的反应,因此具有特殊的临床价值。目前正在测试使用抗体的免疫测定方法,其通过组织切片指示相应的免疫检查点蛋白的存在,以预测对治疗的反应。但是,这些测试仅显示出中等的可靠性。
如前所述,恶性疾病患者的治疗方法在许多情况下并未得到最佳选择,因为通常只能不准确地估计个体患者的病程,因此临床医生没有足够的依据来进行个性化选择或调整可用的治疗方法。因此,缺乏一种可靠且经济的预后和预测方法,其基于客观的测量参数并使得准确地估计疾病的进程以及可靠地预测患有不同恶性肿瘤疾病的患者对于治疗的反应成为可能。
技术实现要素:
在此背景下,根据第一方面,本发明提供了一种用于评估患有恶性疾病的患者的预后和/或用于预测患有恶性疾病的患者对免疫疗法的反应的方法。该方法的特征在于对恶性疾病细胞和/或与恶性疾病t淋巴细胞相互作用的细胞的至少一种免疫调节基因进行dna甲基化分析,并基于该结果确定预后和/或对免疫疗法的反应预测(预估)。
根据第二方面,本发明提供了用于个体化选择用于免疫治疗患有恶性疾病的患者的药物的方法,其中对恶性疾病细胞和/或与恶性疾病t淋巴细胞相互作用的细胞的至少一种免疫调节基因进行dna甲基化分析,并基于该结果选择药物。
本发明的第三方面涉及将对患者的恶性疾病细胞和/或与恶性疾病t淋巴细胞相互作用的细胞的至少一种免疫调节基因进行dna甲基化分析用于预后、预测和/或个性化选择用于该患者的免疫治疗的药物的用途。
本发明的第四方面涉及将恶性疾病细胞和/或与恶性疾病细胞相互作用的t淋巴细胞的免疫调节基因的至少一种cpg-二核苷酸的甲基化的存在、不存在或程度用作预后性和/或预测性生物标记物,或用作针对病人的免疫治疗的个性化选择的生物标记物。
最后,本发明的第五方面提供了用于实施上述的方法或用途的试剂盒。该试剂盒含有至少一个用于dna甲基化分析的寡核苷酸对,在dna中含有的胞嘧啶转化为尿嘧啶或具有与胞嘧啶可区分的碱基配对体和/或分子量的另一种碱基后,寡核苷酸对与来自恶性疾病细胞和/或t淋巴细胞的dna中的免疫调节基因序列杂交,以扩增和/或检测该序列。
在本发明的所有方面,编码免疫检查点的免疫调节基因,所述免疫检查点从包括b7蛋白和它们的受体,mhc:肽复合物结合共同受体,肿瘤坏死因子受体超家族成员tnfrsf9、cd40、tnfrsf4、tnfrsf18和cd27,免疫球蛋白超家族成员tigit、btla、havcr2、btnl2和cd48,以及腺苷结合蛋白腺苷2a受体的组中选择。此外,任意组合的编码所述免疫检查点的免疫调节基因都是可以的。
这些方面的优选变化方案将从说明书和从属权利要求得到。
定义和一般解释
在本说明书中,引用了各种文献,以提供关于本发明的一般技术背景。为避免重复这些文献的公开和教导通过引用完全并入以下说明书中。
以下定义和一般解释旨在指导和帮助本领域技术人员理解,解释和实践本发明。除非另有说明,否则所有技术和科学术语应具有相应于本发明领域的普通技术人员通常的术语理解的那些含义。
本发明的各个方面和变化方案涉及来自分子生物学常规实践的技术和方法。特别地,用于确定cpg二核苷酸的甲基化的dna甲基化分析是分子生物学家或遗传学家的技能之一。这些技术和方法的有用的实验室手册对于本领域技术人员来说是容易获得的,例如,mrgreen和j.sambrook的“molecularcloning,alaboratorymanual”,第4版,2012年,冷泉港实验室出版社。
因此,如本文所用,诸如“一个”或“一个”的不定冠词包括也可能存在这些特征中的两个或更多个的可能性。
如本文所用,术语“恶性疾病”或“恶化疾病”包括具有以下疾病进程特征的疾病,这些疾病具有逐步破坏性并潜在地导致患者的死亡。恶性疾病包括恶性组织生长,例如瘤子或肿瘤,其中恶性肿瘤可通过不受控制的、占据空间的、抑制性的、浸润性和/或侵入性生长来表征。恶性肿瘤通常能够形成继发性肿瘤(转移瘤)。恶性肿瘤包括癌,肉瘤,黑色素瘤,胶质瘤,胚细胞瘤,精原细胞瘤和畸胎瘤。恶性疾病包括血液恶性肿瘤,即影响血液形成系统或造血系统的恶性疾病,如白血病,淋巴瘤,骨髓增生性疾病和骨髓增生异常综合征。白血病包括一组恶性疾病,其中未成熟造血细胞恶性变化,过度增殖并导致细胞在外周血的积聚。淋巴瘤包括淋巴系统细胞退化的疾病。骨髓增生性疾病包括一组疾病,其中一种或多种造血细胞系大量增殖。骨髓增生异常综合征涉及所有造血细胞系的干细胞的克隆扩增,其是造血干细胞的慢性分化障碍的基础。
因此,如本文所用,术语“预后”是指描述患有恶性疾病的患者的健康状况或患者的健康状况将来的变化,其既可包括在没有治疗干预的情况下患者的健康状况,也可以包括患者正在接受或已经接受治疗后的健康状况。本发明意义上的预后还包括患者将来接受治疗后的关于健康状况或健康状态变化的描述。在每种情况下,治疗可以是姑息治疗、治疗性的、新辅助性的或辅助性的。特别地,“预后”包括关于发生的各种健康状况的一种或多种陈述:死亡、存活、复发、淋巴结转移的发生、远处转移的发生、恶性疾病的进展(恶化)、恶性疾病的消退(恢复)、恶性疾病的变化、恶性疾病特异性参数的增加或减少。
术语“预测”应理解为意指恶性疾病对特定疗法的反应的预测,并且对治疗的反应可以通过以下事实来表征:随着疗法的使用,恶性疾病程度逐渐降低,稳定或延缓。反应的缺失可以表征为恶性疾病的程度保持不变,增加或恶化加速。在每种情况下,可以将接受治疗之前的患者或者不接受治疗的患者的恶性疾病的程度作为对照。恶性疾病的程度可以以恶性细胞的数量或恶性肿瘤的大小来表征。特别地,对治疗反应的延迟可以表征为死亡的发生、复发,淋巴结转移的出现、远处转移的出现、恶性疾病的恶化、和/或任何其他恶性疾病特异性参数的增加。
特别地,预后和预测指定与先前的体外方法相关的演绎步骤,使得本发明必需的技术步骤不会发生在人体或动物体上。
如本文所用的术语“基因”是指dna上包含调节,转录和/或功能序列区域的区段,因此含有用于产生生物活性rna的基本信息。特别地,基因包含那些元件诸如启动子、转录因子结合位点、cpg位点、开放染色质、增强子和沉默子、ctcf结合位点,其在基因的转录中实现调节功能。
根据截至2016年4月30日“人类基因组组织基因命名委员会”(hgnc)的建议,指定基因及其核苷酸的命名法。例如,基因家族用斜体拉丁大写字母(例如pdcd1,cd274)表示。
本文所述的基因可通过美国国立卫生研究院的“genbank”公开获得,截至2016年4月30日(bensonda等,nucleicacidsresearch,2013,41,d36-42)。
当以下描述中提及特定dna序列(seqidno)时,其总是包括与所述dna序列具有至少95%、至少96%、至少97%、至少98%或至少99%序列同一性的序列变体。例如,可以用clustalw算法(thompson等,nucleicacidsresearch,1994,22,4673-4680)测定两个核酸序列的序列同一性。
“cpg-二核苷酸”是一种dna基序,其在从5’至3’的通常有效的阅读方向上具有核苷酸序列胞苷-磷酸-鸟苷。鸟苷由核碱基鸟嘌呤和糖β-d-核糖组成。胞苷由核碱基胞嘧啶和糖β-d-核糖组成。
“dna甲基化”是指甲基与dna的特定核苷酸的生物化学偶联或化学偶联。在本发明的上下文中,“dna甲基化”指的是位于cpg-二核苷酸区段中的一个胞嘧啶(5-甲基胞嘧啶)的第五碳原子存在甲基基团。
因此,本发明上下文中的“dna甲基化分析”包括从特定序列环境确定一个cpg-二核苷酸或多个cpg-二核苷酸的甲基化状态。在本发明的各种变体中,“dna甲基化分析”应理解为在胞嘧啶中或在cpg-二核苷酸中是否存在胞嘧啶甲基化。dna甲基化分析可以包含单拷贝的cpg-二核苷酸的。例如,当基因组dna中含有多个细胞的dna时,甲基化分析还可以包括cpg-二核苷酸的多个拷贝。在这种情况下,甲基化分析可以提供甲基化状态或cpg-二核苷酸的甲基化值,即,表示多个拷贝的cpg-二核苷酸中的甲基化程度的横截面值(querschnittswert)。
“免疫检查点”是调节免疫应答的蛋白质,即免疫抑制性的或者是抗炎性(炎症抑制)的或免疫刺激或促炎性(激发炎症)的。免疫检查点用于监测免疫应答的正确功能,例如通过确定抗原特异性t细胞活化的力度和强度或t细胞的效应子的功能是否被共刺激(正调节)或共抑制(负调节)。在某些恶性疾病中,例如在免疫缺失过程中,抗炎性或共抑制免疫检查点被上调。
本发明意义上的“与恶性疾病细胞相互作用的t淋巴细胞”是这些t淋巴细胞,其通过配体-受体结合的方式与恶性疾病的细胞特异性接触或连接。配体可以位于恶性疾病细胞的表面上,例如mhc:肽复合物或mhcii:抗原复合物。受体可以位于t淋巴细胞的表面上,例如t细胞受体(tcr)。或者,配体位于t淋巴细胞的表面上,受体位于恶性疾病细胞的表面上。“与恶性疾病细胞相互作用的t淋巴细胞”,因此也包括这样的t淋巴细胞,其在其所在的位置与抗原呈递细胞接触而活化(激活),可以通过上述的配体-受体结合的方式特异性地与恶性疾病细胞相互作用,甚至不需要先与相应的抗原接触。抗原呈递细胞可以是例如树突细胞或巨噬细胞。例如,激活以t淋巴细胞和来自恶性疾病细胞族抗原中的抗原呈递细胞之间的配体-受体结合的方式发生。配体,例如mhcii:抗原复合物,位于抗原呈递细胞的表面,t细胞受体(tcr)位于t淋巴细胞的表面。在本发明的意义上,t淋巴细胞和恶性疾病细胞之间的另一种相互作用形式包括由恶性疾病细胞产生的腺苷与t淋巴细胞表面上的受体结合。腺苷也可能由t淋巴细胞形成,受体位于恶性疾病的细胞表面。
“免疫疗法”或“免疫治疗”在此用作所有免疫系统的活性受到影响的治疗方法的总称。免疫疗法旨在增强或减轻免疫系统的作用。在本发明的某些变化实施方案中,免疫疗法包括用药物治疗以增强机体对恶性疾病的免疫应答。这些药物包括免疫检查点抑制剂,例如特异性结合免疫检查点并因此阻止其信号传导(特别是抗炎性信号传导)的单克隆抗体。另一种可能性是已经抑制免疫检查点表达的药物,例如通过rna干扰或crispr干扰。然而,药物也可以是免疫检查点激动剂,其增强免疫检查点的信号传导(特别是促炎性信号传导)。
如本文所用,“抑制”免疫检查点意指减缓、抑制或预防一种或多种免疫检查点介导的化学、生物和/或物理性质的反应。
“生物标志物”是特征指标和/或生物学特征,其可以客观地测量和推断生物体中正常生物过程或病理过程的状态,以及正常或患病过程对例如手术、照射或药物处理的干预的反应。生物标志物通常是(生物)化学物质,例如蛋白质,激素,代谢物,糖和核酸,以及其修饰。
“缓和治疗”或姑息治疗被定义为一种药物治疗,其不是以疾病的治愈为目标,而是为了减少缓解症状或其他不良后果(缓解)的医学治疗。因此,它们与以疾病痊愈为目标的治愈性治疗不同。姑息治疗的目的是减缓无法治愈的疾病的进展,减少恶心、疼痛或(反应性)抑郁等症状。
“治愈性治疗”是一种旨在完全恢复患者健康并同时防止其恶化的疗法。特别第,当无法预见痊愈时(机会百分比低)时,也使用该术语。
“新辅助治疗”是一种用于在计划外科手术之前减少肿瘤重量的疗法,通常在肿瘤基本上不能手术时进行,新辅助疗法可减少肿瘤大小,从而使切除肿瘤成为可能。在这种情况下,其通常是实施恶性肿瘤治愈性治疗的唯一方法。
“辅助治疗”是指补充或支持性治疗措施,其在完全去除所有可辨别的肿瘤组织之后应用,以应对可能但尚未检测到的肿瘤转移(微转移),并因此改善长期治愈前景。
前面的一般性描述和以下的详细描述都被认为是示例性的,并且旨在说明要求保护的发明。从以下说明、附图和权利要求中,本发明的其他优点和特征将变得显而易见。尽管将根据本发明的优选实施例阐明本发明,但是在不脱离本发明的范围的情况下可以进行许多其他变化方案。因此,即使没有在权利要求中明确列举,所附权利要求覆盖包括在本发明的真正范围内的特征的变化和组合。
附图说明
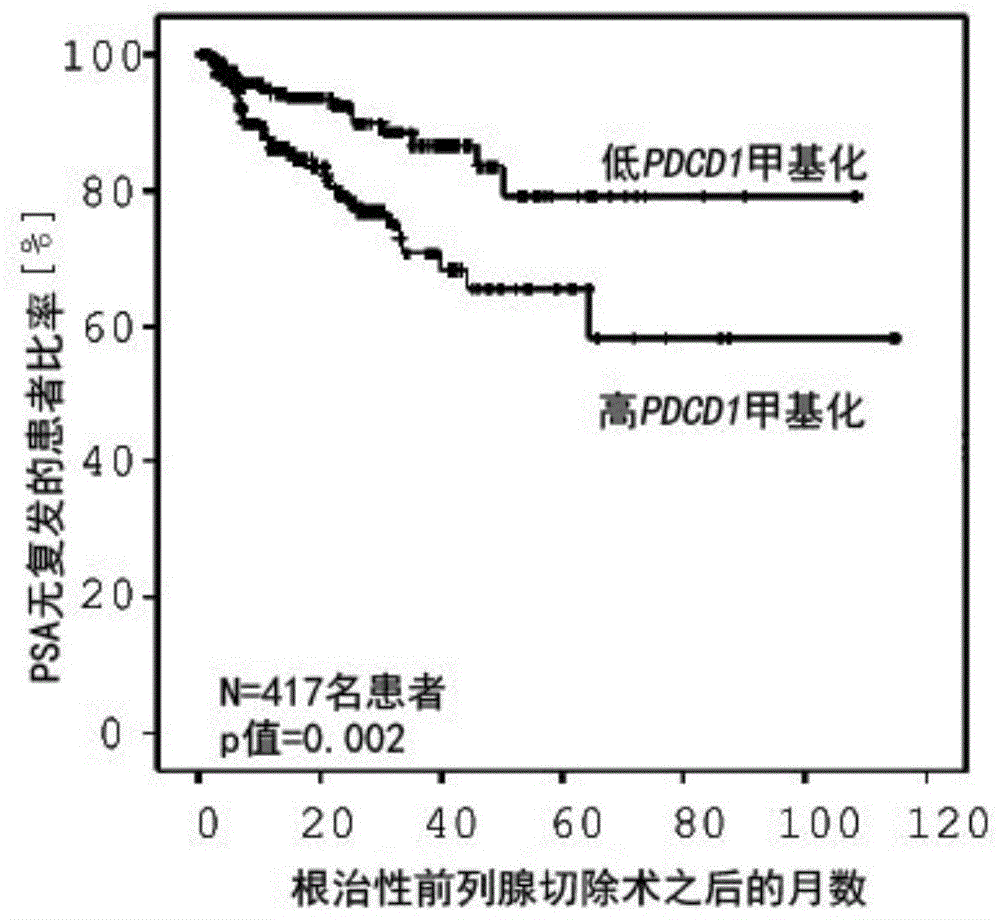
图1显示了根治性前列腺切除术后417名前列腺癌患者的无复发存活的kaplan-meier分析。根据本发明的肿瘤中pdcd1基因位点的dna甲基化分析对患者进行分组。
图2显示了根治性前列腺切除术后417名(a)和259名(b)前列腺癌患者的独立组的无复发存活率的kaplan-meier分析。根据本发明的肿瘤中cd274基因位点的dna甲基化分析对患者进行分组。使用infiniumhumanmethylation450beadchip技术(a),或通过定量甲基化特异性实时pcr(b)进行dna甲基化分析。
图3显示了470名恶性黑素瘤患者的总体存活率的kaplan-meier分析,其使用根据本发明的dna甲基化分析对肿瘤中免疫调节基因pdcd1(a),cd274(b)和pdcd1lg2(c)进行分组。第i组:肿瘤中dna甲基化低于阈值的患者。第ii组:肿瘤中dna甲基化高于阈值的患者。将患者分组的阈值设定为如下的甲基化水平:pdcd1lg2为12.84%;cd274为13.11%,pdcd1为20.54%。
图4显示了470名患有恶性黑素瘤的患者的总体存活的kaplan-meier分析,其通过根据本发明的dna甲基化分析对免疫调节基因tigit(a),tnfrsf9(b),lag3(c),btla(d),ctla4(e)和cd40(f)进行分组。第i组:肿瘤中甲基化低于阈值的患者。第ii组:肿瘤中甲基化高于阈值的患者。将患者分组的阈值设定为以下甲基化水平:tigit为86.41%;tnfrsf9的57.80%;lag3为66.94%;btla占91.31%;ctla4为13.10%;cd40为17.16%。
图5显示了528名头颈部区域鳞状细胞癌患者的总体存活率的kaplan-meier分析,其通过根据本发明的dna甲基化分析对pdcd1基因位点进行分组。患者分组基于所有患者的甲基化中位数,其中一组被指定为高于中位甲基化患者组,另一组被指定为低于中位甲基化患者组。
图6显示对470名患有恶性黑素瘤的患者的研究,根据本发明的基因btla(a)及cd27(c)的dna甲基化分析与mrna表达分析的相关性,以及按照基因btla(b)和cd27(d)的mrna表达进行分组的患者的总体存活率的kaplan-meier分析的结果。
图7显示了对470名恶性黑色素瘤患者的研究,根据本发明的免疫调节基因cd274的mrna表达分析与dna甲基化分析(a)的相关性,以及根据本发明的dna甲基化分析(b)、mrna表达分析(c)以及mrna表达分析与dna甲基化分析的组合(d)进行分组的患者的总体存活率的kaplan-meier分析的结果。
图8显示了470名患有恶性黑素瘤的患者的总体存活率的kaplan-meier分析,其通过根据本发明的免疫调节基因cd274(a)(如图7中),pdcd1lg2(b),pdcd1(c),btla(d),lag3(e),tigit(f),cd40(g),ctla4(h)和tnfrsf9(i)的dna甲基化分析和mrna表达分析而分组。第ii组(高风险组):高dna甲基化和低mrna表达;第iv组(低风险组):低甲基化和高mrna表达;第i+iii组(中等风险组):高mrna表达和高dna甲基化或低mrna表达和低dna甲基化。
图9显示了根据本发明的470名患有恶性黑素瘤的患者的总体存活率的kaplan-meier分析,其按照9个免疫调节基因cd274,pdcd1,pdcd1lg2,tigit,tnfrsf9,lag3,btla,ctla4和cd40的mrna表达分析(a),dna甲基化分析(b)和组合的mrna表达分析和dna甲基化分析(c)进行分组。第i组:9个基因中至少8个mrna表达升高的患者;第ii组:9个基因中少于8个mrna表达增加的患者;第iii组:九个基因中最多四个dna甲基化增加的患者;第iv组:九个基因中至少五个dna甲基化增加的患者;第v组:属于第i组和第iii组的患者;第vi组:属于第ii组和第iv组的患者;第vii组:属于第i组和第iv组或第ii和iii组的患者。
图10显示免疫调节基因lag3的mrna表达与启动子区(a)和基因内区(b)的dna甲基化的相关性;免疫调节基因cd40的mrna表达与启动子区域(c)和基因内区域的(d)的dna甲基化的相关性;以及470名恶性黑色素瘤患者的整体存活率的kaplan-meier分析,其按照本发明免疫调节基因lag3(e,f,g)和cd40(h,i,j)的启动子(e,h)、内区域(f,i),和上述两个区域组合(g,j)的dna甲基化分析进行分组。第i组和第iv组:dna甲基化低于阈值;第ii组和第iii组:dna甲基化高于阈值;第v组:ii组和iv组的患者;第vi组:ii组和iii组或i组和iv组的患者;第vii组:第i组和第iii组的患者。
图11显示了182名急性髓系白血病总体存活率的kaplan-meier分析,其中患者按照本发明免疫调节基因cd274(a,b,c),pdcd1(d,e,f)和pdcd1lg2(g,h,i)的dna甲基化分析(a,d,g)、mrna的表达分析(b,e,h),以及mrna表达分析和dna甲基化分析的组合(c,f,i)分组。第i组:dna甲基化低于阈值;第ii组:dna甲基化高于阈值;第iii组:mrna表达高于阈值;第iv组:mrna表达低于阈值。阈值:cd274的dna甲基化为17.00%;pdcd1的dna甲基化为56.95%;pdcd1lg2的dna甲基化为62.82%;cd274的mrna表达为4.7319;pdcd1的mrna表达为23.7933,pdcd1lg2的mrna表达为12.9895。
图12显示了182名急性髓系白血病总体存活率的kaplan-meier分析,其中患者按照本发明的免疫调节基因tnfrsf9(a),tigit(b),btla(c),havcr2(d),cd80(e),ctla4(f),icos(g),c10orf54(h),hhla2(i),cd160(j),kir2dl4(k)和kir3dl1(l)的dna甲基化分析进行分组。二分法是基于优化的阈值。第i组:dna甲基化低于该阈值;第ii组:dna甲基化高于阈值。
图13显示了510名低级别胶质瘤患者总体存活率的kaplan-meier分析,患者按照本发明的免疫调节基因cd274(a,b,c),pdcd1(d,e,f)和pdcd1lg2(g,h,i)的dna甲基化分析(a,d,g)、mrna的表达分析(b,e,h)和组合的dna甲基化分析和mrna表达分析(c,f,i)进行分组。以下阈值用于dna甲基化的二分法:cd274为32.00%;pdcd1为33.79%,pdcd1lg2为59.89%。mrna表达的二分法基于来自患者组的所有肿瘤的相应基因的mrna表达的中位数。第i组:低于阈值的dna甲基化;第ii组:高于阈值的dna甲基化;第iii组:mrna表达高于中位数;第iv组:mrna表达低于中位数;第i组+第iii组:第i组和第iii组的患者;第ii+iv组:第ii组和第iv组的患者;第i组+第iv/ii+iii组:第ii组和第iii组或i和iv组的患者。
图14显示了510例低级别胶质瘤患者总体存活率的kaplan-meier分析,患者根据本发明的免疫调节基因cd80(a,b,c),ctla4(d,e,f),icos(g,h,i),cd276(j,k,l)的dna甲基化分析(a,d,g,j)、mrna的表达分析(b,e,h,k),组合的dna甲基化分析和mrna表达分析(c,f,i,l)进行分组。对于基于dna甲基化的二分法,选择以下阈值:90.76%(cd80);6.43%(ctla4);84.75%(icos)和30.08%(cd276)。mrna表达的二分法基于来自患者组的所有肿瘤的相应基因的mrna表达的中位数。第i组:低于阈值的dna甲基化;第ii组:高于阈值的dna甲基化;第iii组:mrna表达高于中位数;第iv组:低于中位数的mrna表达;第i组+第iii组:第i组和第iii组的患者;第ii+iv组:第ii组和第iv组的患者;第i组+iv/ii+iii组:第ii组和第iii组或i和iv组的患者。
图15显示了510例低级别胶质瘤患者总体存活率的kaplan-meier分析,患者根据本发明的基因tnfrsf25(a),tnfrsf9(b),cd40(c),tigit(d),btla(e),havcr2(f),c10orf54(g),hhla2(h),lag3(i),cd160(j),kir2dl4(k)和kir3dl1(l)的dna甲基化分析进行分组。二分法基于以下阈值:tnfrsf25为61.88%;tnfrsf9为79.24%;cd40为43.24%;tigit为80.66%;btla为84.64%;havcr2为2.495%;c10orf54为66.52%;hhla2为77.14%;lag3为74.34%;cd160为92.89%;kir2dl4为90.85%,kir3dl1为52.28%。第i组:低于阈值的甲基化;第ii组:高于阈值的甲基化。
图16显示了318例透明细胞肾细胞癌患者的整体存活率的kaplan-meier分析,根据本发明的免疫调节基因tigit(a),tnfrsf9(b),cd274(c),cd80(d),ctla4(e),cd276(f)和hhla2(g)的dna甲基化分析进行分组。二分法基于来自所有患者的肿瘤相关基因的dna甲基化的中位数。第i组:甲基化低于中位数;第ii组:甲基化高于中位数。
图17显示了318例透明细胞肾细胞癌患者的整体存活率的kaplan-meier分析,其根据本发明的免疫调节基因tigit(a),tnfrsf9(b),cd80(c),ctla4(d),cd276(e)和hhla2(f)的dna甲基化分析和mrna表达分析的组合进行分组。二分法基于来自所有患者的肿瘤的相应基因的dna甲基化或mrna的中位数。第i组:dna甲基化低于中位数,mrna表达高于中位数;第iii组:dna甲基化高于中位数,mrna表达低于中位数;第ii组:dna甲基化高于中位数,mrna表达高于中位数或者dna甲基化低于中位数,mrna表达低于中位数。
图18显示了本发明免疫调节基因cd274的dna甲基化分析,用于确定患者对于使用药物帕母单抗(pembrolizumab)的免疫疗法的反应,其中免疫调节活性通过基因pdcd1所编码的免疫检查位点而受到抑制。显示了23名在免疫疗法开始之前通过外科手术切除恶性黑色素瘤的患者的转移瘤中该基因的甲基化值(%)。
图19示显示了根据本发明免疫调节基因pdcd1的dna甲基化分析,用于确定患者对于使用药物帕母单抗的免疫疗法的反应,其中免疫调节活性通过基因pdcd1所编码的免疫检查位点而受到抑制。显示了23名在免疫疗法开始之前通过外科手术切除恶性黑色素瘤患者的转移瘤中该基因的甲基化值(%)。
序列说明
seqidno:1到seqidno:485分别在序列表中的数字序号<213>或<223>中给出。
具体实施方式
本发明的第一方面涉及一种用于评估恶性疾病患者的预后和/或用于预测患者对恶性疾病免疫疗法的反应的方法。在这种情况下,对恶性疾病细胞和/或与恶性疾病的细胞相互作用的t淋巴细胞的至少一种免疫调节基因进行dna甲基化分析。此处的免疫调节基因编码选自一个从以下组中选择的免疫检查位点:b7蛋白及其受体,即b7蛋白作为配体结合的受体,mhc:肽复合物结合共同受体,肿瘤坏死因子受体超家族成员tnfrsf9,cd40,tnfrsf4,tnfrsf18,和cd27,免疫球蛋白超家族成员tigit,btla,havcr2,btnl2和cd48以及腺苷结合腺苷2a受体。另外也可以对多个免疫调节基因进行dna甲基化分析,其可以包括这些免疫检查位点的任意组合。优选地,在这种情况下,对涉及相同配体-受体结合的几种免疫调节基因进行dna甲基化分析。基于dna甲基化分析的结果,然后确定预后和/或预测对免疫疗法的反应(预测)。
本发明基于以下认知作出,恶性疾病具有复杂的遗传和表观遗传变化,因此可以是高度个性化的。因此,即使是相同器官和相同阶段的恶性疾病也可导致患者的不同预后。发明者在其研究中已经意识到恶性疾病具有免疫学亚型,其被身体的免疫系统更好或更差的维持,一般会导致更为有利的或更不利的疾病过程。这个属性可以归因于一个事实,即这些亚型具有免疫调节基因的dna甲基化和mrna表达的特定特征。
根据本发明人的研究,在具有有利结果的患者中,恶性疾病细胞或与恶性疾病细胞相互作用的t淋巴细胞或这两种细胞类型中,一个或多个b7-蛋白和它们的受体、一个或多个mhc:肽复合物结合共同受体、一个或多个tnfrsf9,cd40,tnfrsf4,tnfrsf18和cd27,一个或多个tigit,btlahavcr2,btnl2,cd48和/或腺苷结合腺苷2a受体特征性表达。发明者能够在这方面首次证明这些免疫调节基因的表达受甲基化调节。以这种方式可以通过相应的dna甲基化分析来描述免疫学表型,所述免疫学表型通过免疫调节基因的甲基化来定义和客观测量。
在本发明的上下文中,已发现免疫调节基因的甲基化与患者的恶性疾病的预后显著相关。本发明人的一个重要且极其令人惊讶的发现是它是恶性疾病的一般现象,其不限于特定实体。因此,本发明第一次提供了预后测试,即基于相同基因的dna甲基化分析为各种恶性疾病类型的疾病进展提供具有普遍适应性的预测,而基于先前已知的作为生物标志物的基因的甲基化分析仅涉及到单个或少数实体的一种预测。在这方面,还参考以下实施方案。
本发明的另一个独特的特征和优点是由发明人所鉴定的免疫调节基因,其的dna甲基化是恶性疾病的进展的预后,同时也编码免疫检查位点,其代表免疫疗法的主要目标。因此了解其相应的基因产物的存在或不存在的程度与规划有效的免疫疗法密切相关。因为已经表明免疫调节基因的dna甲基化与相应的免疫检查点的表达直接相关,本发明方法可以替代或补充预后的评估和患者对适当免疫疗法的反应的预测。例如,如果dna甲基化分析指明免疫检查点的表达,则很可能有反应。因此,首次地,在同一测试中,在确定患者预后的同时预测对免疫治疗的反应,而基于现有生物标记基因的传统方法目前为止只能或者进行预后或者进行预测。
dna甲基化分析基本上可以用所有常规方法进行,这些方法在本领域相关文献是已知的。合适的方法包括,例如,以下步骤:a)分别提供恶性细胞或t淋巴细胞的dna;b)将dna中包含的至少一部分胞嘧啶转化为尿嘧啶或具有与胞嘧啶可区分的碱基配对体和/或分子量的另一碱基;c)用步骤b)获得的dna检查免疫调节基因的dna甲基化。
在步骤a)中分析的dna可以来自各种来源,例如从手术或活检获得的组织浸润性t淋巴细胞和恶性疾病的细胞。细胞也可以来自涂片和抽吸物,例如冲洗液,细针抽吸物或痰液。所述dna也可从血液、血清、血浆中获得,例如从游离循环的dna、外来体dna、或以自由循环细胞的形式中回收dna。所述dna也可以从自其他体液,例如尿液、胸腔积液或腹水,例如可以从其中游离形式的dna或细胞回收dna。dna也可以从未经保存的(新鲜的)细胞、组织和体液以及固定的细胞、组织和体液中获得。细胞、组织和体液的固定可以通过沉淀诸如乙醇和其他醇的固定剂或通过交联剂诸如甲醛的固定剂来实现。dna也可以来自这些来源的任何组合。它也可以从上述来源提取dna。还可以富集dna,例如通过沉淀或提取。例如,这可以从所提及的体液中自由循环的dna提供。还可以富集细胞,例如通过尺寸过滤或通过表面上携带抗体的磁性颗粒,其抗原位于待富集的细胞表面上。例如,这可以在恶性疾病的自由循环细胞或来自所述体液的t淋巴细胞中提供。待分析的dna的其他合适来源是新鲜组织的匀浆和固定组织的裂解物。
在一个优选的变型方案中,dna包括自由循环的dna、来自外来体的dna和/或来自体液自由循环细胞的dna,即所谓液体活检或“液体活检”。液体活检目前代表肿瘤学研究的一个主要领域,其不分析可疑组织本身,因此,例如肿瘤组织,而是分析体液的样品,例如血液样品。在这些样品中,可以分析不同的物质,其包括来自肿瘤的、自由循环的基因dna、外来体dna、或从肿瘤中释放进血管中的自由循环的细胞。当肿瘤或其转移无法进行活检或如果患者在癌症晚期活检将构成太大的威胁的情况,则根据本发明的方法有利地使进行体液活检分析。本发明的优势在于该免疫调节基因的dna甲基化在体液中是很好测量的,而传统的基于mrna或免疫组织化学的免疫调节基因的表达检测是困难的或者甚至是不可能的。
原则上,步骤b)中dna的转化可以用本领域已知的所有合适的方法进行。通常,它涉及化学或酶促转化,例如通过使dna与例如亚硫酸氢钠或亚硫酸氢铵的亚硫酸氢盐接触。
如果需要,在步骤b)中的转化之后和步骤c)中的dna甲基化检查之前,可以纯化dna。合适的纯化方法和方案是本领域技术人员已知的,并且可包括例如dna提取,沉淀或聚合物介导的富集(polymer-mediatedenrichment)。
dna甲基化分析可包括确定免疫调节基因中包含的至少一个cpg二核苷酸的甲基化的存在、不存在或程度。还可以检查免疫调节基因的多个cpg二核苷酸。这些也可以分布在待检查的免疫调节基因的不同部分上。在优选的变化方案中,要确定至少两种不同免疫调节基因的至少一个cpg二核苷酸的甲基化。以这种方式,本发明解决了恶性疾病的dna中的cpg二核苷酸有时有异质甲基化的问题。因此可以避免假阴性和假阳性试验结果,从而实现了特别可靠并尤其特别具有区分性的预后和/或预测,如以下实施方案所清楚地显示的。
步骤c)中的免疫调节基因的dna甲基化的检查不受任何特定限制。基于本公开内容,本领域技术人员可以容易地确定合适的方法。在这方面,还参考上述实验室手册。在优选变型中,首先进行聚合酶链反应(pcr),即用寡核苷酸,所谓的引物,将转换的dna进行扩增,其包含至少一个待分析的cpg二核苷酸。优选地,随后至少对扩增产物的一部分进行测序,例如sanger测序、焦磷酸测序、质谱测序或测序的第二或第三代,这称为“高通量并行测序”、“下一代测序(ngs)”或称为纳米多孔测序。也可以在pcr后与突变特异性寡核苷酸(探针)进行杂交,例如以dna微阵列的形式。甲基化也可以通过定量实时pcr(quantitativereal-timepcr,qpcr)确定,如果合适的话,通过熔解曲线分析。特别是,定量实时pcr可以用如wo1997/046705a1中所描述的甲基化特异性引物和/或如wo2002/072880a2中所描述的甲基化特异性阻断寡核苷酸来处理。在一个优选的变型方案中可以使用甲基化特异性检测探针。
在其它优选的变化方案中,pcr可被取消,比如“全基因组鸟枪硫酸氢盐测序”(wgsbs)或直接纳米孔测序。在wgsbs中dna被片段化,然后通过衔接子连接到dna的片段上。扩增和测序完成之后使用衔接子是可能的。另外,也可以在wgsbs省略片段化的步骤,因为dna在例如亚硫酸氢盐的处理中已经被碎片化。用于实施wgsbs的步骤是本领域技术人员容易获得的(johnson,m.d.etal.,curr.protoc.mol.biol.,2012,99,21.23.1-21.23.28;lister,r.etal.,nature,2009,462,315-322;berman,b.p.etal.,nat.genet.,2011,44,40-46)。
在另一个优选的变型中,可以在pcr扩增之前进行特定寡核苷酸(探针)的杂交,其在结合的情况下连接并随后通过pcr进行扩增。本领域技术人员可以没有任何问题地获取适当的方法和步骤,诸如“多重连接依赖性探针扩增”(mlpa)。例如,b.d.m.theophilus和r.rapley所著的“pcr突变检测方法”,第2版,2011,斯普林格。
在进一步优选的变型中,使用所述的无限人类甲基化450珠芯片(infiniumhumanmethylation450beadchip)进行甲基化分析。例如在章节determinationofdnamethylationlevelsusingilluminahumanmethylation450beadchips(利用“无限人类甲基化450珠芯片确定dna甲基化水平)vonm.a.carless,welchesimbuch,,chromatinprotocols(《染色质步骤》)“vons.p.chellappan,band1288,2015,derbuchserie,,methodsinmolecularbiology(《分子生物学方法》)“,springerscience+businessmedianewyork中可以找到合适的方案。其他合适的方案在以下实施方案中是显而易见的。
dna甲基化分析优选在允许定量测定至少一个cpg二核苷酸的甲基化的条件下进行。定量测定还可以包括一个或多个基因中多个cpg二核苷酸的甲基化状态。然后将所获得的测量值平均化,例如以便获得特别稳定的值。以这种方式,实现了该方法的特别高的可靠性和精确性。定量甲基化分析例如适用于确定相对甲基化,通过将甲基化基因位点的拷贝数与同一基因位点的拷贝总数相关联。
然后例如可以将dna甲基化分析或免疫调节基因的甲基化的结果和参考值进行比较,以确定预后和/或患者的对于免疫疗法的可能反应。设定适当的参考值是实验室医疗程序的一部分。一种可能是,一组患者具有可对比的恶性疾病,特别是相同的实体,其预后和/或对于免疫疗法的反应在回顾性分析中是已经公知的,对其进行免疫调节基因的dna甲基化分析。在此基础上,所述的免疫调节基因的dna甲基化可以与预后和/或对免疫疗法的可能反应相关联。例如,回顾性分析组划分成两个或更多亚组,预后或反应可通过免疫调节基因的dna甲基化来区分。那么合适的参考值,例如是dna甲基化值,其划分两个亚组,以下也称作为阈值。因此根据dna甲基化分析的结果是高于还是低于根据本发明方法得到的阈值,则可以将个体患者分归入具有已知预后或已知反应的组中的一个。
还可以将dna甲基化分析的结果与免疫调节基因的mrna表达相关联。例如,如果该基因的表达是患者预后的指标,那么可以通过基于该基因的dna甲基化确定mrna表达来确定患者的预后。由于根据本发明的免疫调节基因的表达还提供了用于免疫治疗性的靶标,因此可以以这种方式可靠地预测对相应免疫疗法的反应,其使用由免疫调节基因编码的免疫检查点作为靶标。
本发明的特征尤其在于本发明的免疫调节基因的dna甲基化分析对于许多不同肿瘤类型中疾病过程的预后或对于免疫疗法的反应的预测是通用的,这对本领域技术人员来说是不曾预见的。恶性疾病可以特别地包括癌,黑素瘤,肉瘤,神经胶质瘤,淋巴瘤和/或白血病。癌症可以例如是腺癌,鳞状细胞癌,小细胞癌,神经内分泌癌,肾细胞癌,移行细胞癌,肝细胞癌,肛门癌,肺癌,子宫内膜癌,胆管细胞癌,肝细胞癌,睾丸癌,结直肠癌,头部癌颈部,包括食道,胃癌,乳腺癌,肾癌,卵巢癌,胰腺癌,前列腺癌,甲状腺癌和/或子宫颈癌。甲肉瘤可以例如是血管肉瘤,软骨肉瘤,尤因肉瘤,纤维肉瘤,卡波西氏肉瘤,脂肪肉瘤,平滑肌肉瘤,恶性纤维组织细胞瘤,神经源性肉瘤,骨肉瘤或横纹肌肉瘤。白血病例如是急性骨髓性白血病(aml),急性淋巴细胞性白血病(all),慢性淋巴细胞性白血病(cll),或慢性髓细胞白血病(cml)。淋巴瘤可以是霍奇金淋巴瘤或非霍奇金淋巴瘤。非霍奇金淋巴瘤可以是b细胞淋巴瘤或t细胞淋巴瘤。在实施方案中可以找到相应的说明。
在该方法的另一个变型中,除了dna甲基化分析之外,还进行免疫调节基因的mrna表达分析。这可以包括,例如,确定免疫调节基因的至少一种转录变体或至少两种不同免疫调节基因的至少一种转录变体的mrna表达水平。优选地,mrna在允许定量测定mrna表达水平的条件下表达分析。
用于mrna表达分析的合适方案是本领域技术人员公知的,并且通常包括以下步骤:a)提供恶性细胞或t淋巴细胞的mrna;b)将mrna转换成cdna;c)检测免疫调节基因的cdna。然而,也可以直接分析mrna而无需事先转换成cdna。特别地,纳米技术适用于此目的。所述的mrna表达分析也可以通过rna-seq(rna测序),或全转录组鸟枪法测序(wtss)的手段来执行。这里为了确定mrna的定性或和/或定量可以使用下一代测序(ngs)方法。例如在实施方案中,可确定一个特定序列的mrna分子的数量,并且与所有的mrna分子的数量相关联,以确定归一化数(归一化计数)。合适的rna测序技术和解决方案可例如从illumina公司,sandiego,ca,usa处获得。
令人惊讶地,根据本发明的dna甲基化分析和mrna表达分析的组合导致预后评估的进一步改进,在这个意义上,预对测组的更精确的分组是可能的。所述分析组合使得在每种情况下既可以基于免疫调节基因的dna甲基化分析也可以基于免疫调节基因的mrna表达分析为患者确定预后,例如基于上述参考值。随后,可以将这些预后组合在一起,以便进一步将预后分为亚组。例如,如果免疫调节基因的mrna表达分析以及dna甲基化分析后预后都很差,则可以形成预后不良的亚组。例如,如果免疫调节基因的mrna表达分析以及dna甲基化分析后预后都很好,则可以形成预后优良的亚组。例如,两个中间预后组包括那些对免疫调节基因的dna甲基化分析或mrna表达分析表明预后良好而另一分析表明预后不良的患者。如果分析几种免疫调节基因,则可以进行更加精细的分组,从而可以进行预后组的多因素分类。在此基础上,借助于本发明可以基于客观可测量的参数,以前所未有的方式进行临床决策,从而更准确、更可靠地与个体患者匹配。
恶性疾病细胞的dna甲基化的改变涉及基因内和周围的各种cpg二核苷酸。对于根据本发明的方法,cpg二核苷酸特别适用于启动子和恶性疾病或t淋巴细胞的免疫调节基因编码的转录物的序列。然而,恶性疾病细胞或t淋巴细胞的免疫调节基因外部的cpg二核苷酸的甲基化也可以发生变化,也适合于本发明的方法。根据本发明的dna甲基化分析的优选区域包括调节基因区域,特别是转录因子结合位点,启动子,cpg位点,沉默子,增强子,ctcf结合位点及它们的组合。例如远离基因的增强子称作远端增强子。增强子也可位于基因附近,因此命名为近端增强子。调节基因区域是本领域技术人员公知的,例如在d.s.latchman,第二版,2015,garylandscience,taylor&francisgroup,llc中描述的“基因调控”。根据本发明的方法的实施方案,每个cpg-二核苷酸的甲基化水平与基因的表达或转录活性相关。例如,转录活性借助染色体结构改变而是明显的。所谓“开放染色质”可能与基因的高转录活性相关,如在genetik(基因组学)“vonw.janning和e.kunst,2004,georgthiemeverlag,stuttgartundnewyork中所描述。术语“开放染色质”因此适用于本发明的dna甲基化分析。
本领域技术人员基于合适的数据库而无需其它就可以容易地确定调控基因元件。例如,在数据库中的这种调控元件以“ensembl”注明,如在theensemblregulatorybuild“vond.r.zerbino,s.p.wilder,n.johnson,t.juettemannundp.r.flicek,2015,genomebiology(基因组生物学),ausgabe16,doi:10.1186/s13059-015-0621-5中所述。
合适的人类基因组的一级序列,借助于其的合适的和优选的范围和序列也能够用于根据本发明免疫调节基因的dna甲基化分析,例如,人类基因组序列第38版(genomereferenceconsortiumhumanbuild38,grch38)或2016年3月21日发表人类基因组序列第38版释放补丁7(grch38.p7)。基因组的区域按照下列方式书写:“染色体序号:区域内第一个碱基的位置-区域内最后一个碱基的位置”,也就是例如,“2:241849881-241858908”为第2染色体中碱基241849881到碱基241858908的区域。
在一个变体中,免疫调节基因选自编码b7蛋白及其受体的基因cd274,pdcd1lg2和pdcd1。在进一步的变型中,所述免疫调节基因是从编码b7蛋白及其受体的基因cd80和ctla4中选择。在又一变型中,所述免疫调节基因是从编码b7蛋白受体的icos基因和编码b7蛋白的基因cd276,c10orf54和hhla2中选择。在又一变型中,所述免疫调节基因是从编码mhc:肽复合物结合共受体的基因lag3,cd160,kir2dl4,kir3dl1中选择。来自这些变体的免疫调节基因的任何组合也是可能的。
基因pdcd1或程序性细胞死亡蛋白1也是cd279,pd1,hpd-1,sleb2,pd-1,hsle1和hpd-l的同义名,这是已知的。pdcd1是免疫调节基因并编码本发明意义上的免疫检查点的蛋白。蛋白pdcd1是受体,并且可以结合例如属于b7蛋白的两个配体,其由基因cd274和pdcd1lg2编码。pdcd1属于免疫球蛋白超家族。pdcd1的dna甲基化分析的优选范围包括:编码转录区域(2:241849881-241858908,seqidno:17),开放染色质区域(2:241849051-241853001,seqidno:18和2:241861820-241862593,seqidno:19),增强子(2:241852997-241855201,seqidno:2),一个ctcf结合位点(2:241859081-241860074,seqidno:3)和/或启动子(2:241856912-241861429,seqidno:29和2:241862929-241865230,seqidno:30)。pdcd1的dna甲基化分析的更优选范围选自那些在实施例1,3,5,7,8,10,12和17中为此目的描述的那些。对于根据本发明的mrna检测的表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于seqidno:73,seqidno:74和/或seqidno:75。特别地,对免疫治疗的反应的预测包括适于抑制pdcd1编码的受体和/或其配体的药物,其中,该配体由免疫调节基因pdcd1lg2和cd274编码。例如nivolumab(商品名:opdivo,由bristol-myerssquibb公司制造),帕母单抗(商品名:keytruda,由merck/msdsharp&dohme公司制造),pidilizumab(ct-011,由curetechltd.制造),mgd013(macrogenics),amp-224(由葛兰素史克制造),medi0680(amp-514制造商:medimmunellc),aunp-12(由aurigenediscoverytechnologiesltd.制造),bms935559(mdx1105,由bristol-myerssquibb制造),ca-170(由curisinc.制造),mpdl3280a(由roche制造),medi4736(由astrazeneca制造),avelumab(msb0010718c,由pfizer制造)和rhigm12b7(b7-dc交联抗体rhigm12b7,梅奥诊所)。
基因cd274或cd274分子也是pdcd1l1,b7-h,b7-h1,pdcd1lg1,pd-l1,pdl1和b7h1的同义名,这是已知的。cd274是免疫调节基因并编码b7蛋白,其是本发明意义上的免疫检查点。cd274编码通过pdcd1编码的受体的配体的蛋白。cd274属于免疫球蛋白超家族。cd274的dna甲基化分析的优选范围包括:编码转录区域(9:5450503-5470566,seqidno:1)和启动子(9:5445402-5456799,seqidno:76和9:5458041-5461360,seqidno:77),增强子(9:5457122-5457702,seqidno:78;9:5463574-5468340,seqidno:79;9:5440647-5441785,seqidno:80和9:5472191-5473149,seqidno:81)和/或ctcf结合位点(9:5440970-5441435,seqidno:82和9:5446325-5446870,seqidno:83)。dna甲基化分析的更优选范围选自cd274的编码区域,其包括可变启动子和一个ctcf-结合位点(9:5451072-5481072,seqidno:371),可变启动子(9:5451072-5461819,seqidno:375),该启动子的上游调节区(9:5433159-5449887,seqidno:370),启动子上游的区域,其包含一个ctcf结合位点(9:5439290-5449887,seqidno:376),启动子的下游编码区(9:5451072-5470566,seqidno:372),以及编码区下游的区域,其中包含一个ctcf结合位点和一个增强子(9:5470566-5496357,seqidno:373)。对于cd274的dna甲基化分析更优选的范围选自在实施例2,3,7,8,10,12,15和17中为此目的描述的那些。对于本发明的mrna检测的表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:28,seqidno:101,seqidno:102,seqidno:103和/或seqidno:104。特别地,对免疫治疗的反应的预测包括适于抑制cd274编码的b7蛋白和/或相应的pdcd1编码的受体和/或pdcd1lg2编码的配体(其同样与pdcd1编码的受体结合)的药物。例如nivolumab,帕母单抗,pidilizumab,amp-224,amp-514,aunp-12,bms935559,ca-170(curis),atezolizumab(mpdl3280a,roche,genentech),mgd013(macrogenics),medi4736,ca-170(curis,inc.)和avelumab。
基因pdcd1lg2或程序性细胞死亡蛋白1配体2也是btdc,pdl2,cd273,pdcd1l2,b7-dc,ba574f11.2,pd-l2和b7dc的同义名,这是已知的。pdcd1lg2是免疫调节基因,其编码b7蛋白,这是本发明的意义上的免疫检查点。pdcd1lg2编码b7蛋白,其是通过pdcd1编码的受体的配体。pdcd1lg2属于免疫球蛋白超家族。用于pdcd1lg2的dna甲基化分析的优选范围包括:编码转录区域(9:5510570-5571254),以及启动子(9:5507688-5523442,seqidno:84;9:5491444-5503289,seqidno:85;9:5528150-5534251,seqidno:86和9:5547972-5571492,seqidno:87)和/或增强子(9:5479110-5491616,seqidno:88;9:5522642-5528253,seqidno:89;9:5534822-5547690,seqidno:90;9:5572730-5580962seqidno:91),以及该编码序列的上游区域中(9:5496357-5510570,seqidno:374)。特别优选地,一个或多个cpg二核苷酸的dna甲基化分析位于序列区域9:5433159-5603325,其编码相邻的基因cd274和pdcd1lg2,并包括这些基因的增强子和启动子,尤其是,cd274和pdcd1lg2的编码区域之间的区域(9:5470566-5510570;seqidno:485)。用于pdcd1lg2的甲基化分析更优选的范围是选自在实施例3,5,7,8,10和13中为此目的描述的那些。对于根据本发明的mrna检测的表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于seqidno:105。特别地,对免疫治疗的反应的预测包括能抑制由pdcd1lg2编码的b7-蛋白和/或通过pdcd1编码的相应受体的药物。合适的抑制剂例如是nivolumab,帕母单抗,pidilizumab,amp-224,amp-514,aunp-12,ca-170(curis,inc。)和rhigm12b7。
基因icos或诱导性t细胞共刺激子也具有同义名cd278,cvid1和ailim,这是已知的。icos是一种免疫调节基因,编码一种是本发明意义上的免疫检查点的蛋白质。icos是一种受体,可以和b7蛋白所属的配体结合,其通过基因icoslg编码。icoslg或诱导性t细胞共刺激配体是免疫调节基因并编码配体,所述配体是在本发明的意义上的免疫检查点。icoslg也称为icosl,b7rp1,licos,b7rp-1,kiaa0653,gl50,icos-l,cd275,b7h2和b7-h2。icos和icoslg属于免疫球蛋白超家族。用于icos的dna甲基化分析的优选范围包括以下区域:转录编码区域(2:203936748-203961577,seqidno:92),以及启动子(2:203934590-203941036,seqidno:93和2:203948548-203953636,seqidno:94)和/或增强子(2:203931099-203937863,seqidno:95和2:203940518-203949061,seqidno:96)。用于icoslg的dna甲基化分析的优选范围包括:转录编码区域(21:44222991-44240966,seqidno:97)以及cpg-岛(21:44240132-44243380,seqidno:98),启动子(21:44238919-44247988,seqidno:99)和/或增强子区域(21:44214476-44227512,seqidno:100)。icos和icoslg的dna甲基化分析进一步优选的范围选自在实施例11,13和17中为此目的描述的那些。对于根据本发明的icos或icoslg的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于seqidno:106,seqidno:107,seqidno:108,seqidno:109,seqidno:110和/或seqidno:111。一种优选的方法实施方案包括使用icos和/或icoslg基因的dna甲基化分析来确定对药物的反应,该药物能够改变icos和/或icoslg的免疫调节作用,特别是激活由icos编码的受体,合适的活化剂是例如jtx-2011(jouncetherapeutics)。
基因ctla4或细胞毒性t淋巴细胞的相关蛋白4也具有同义名celiac3,grd4,ctla-4,iddm12,cd,cd28,gse,cd152和alps5,这是已知的。ctla4是免疫调节基因,并编码一种本发明的意义上的免疫检查点的蛋白。ctla4编码例如能够与属于b7蛋白配体结合的受体,该配体由基因cd80编码。cd80或cd80分子是免疫调节基因,其编码的配体是在本发明的意义上的免疫检查点。cd80例如也称为b7,cd28lg1,b7.1,b7-1,lab7,bb1和cd28lg。ctla4和cd80属于免疫球蛋白超家族。用于ctla4的甲基化分析的优选范围包括:转录编码区(2:203867786-203873960,seqidno:160),以及启动子(2:203866174-203868926,seqidno:162和2:203869477-203874095,seqidno:164),增强子(2:203874152-203875266,seqidno:165;2:203876672-203878051,seqidno:166和2:203879313-203881585,seqidno:167)和/或ctla4和相邻免疫调节基因icos之间的区域(2:203872383-203939876)。用于ctla4的dna甲基化分析更优选的范围选自在实施例4,7,8,11,13,15和16中为此目的描述的那些。用于cd80的dna甲基化分析优选范围例如包括:转录编码区(3:119524293-119559602)和启动子(3:119554042-119563668,seqidno:163和3:119568227-119573274,seqidno:177)和/或增强子(3:119563379-119568778,seqidno:178;3:119538188-119543511,seqidno:179和3:119545840-119554325,seqidno:180)。用于cd80的甲基化分析更优选的范围选自在实施例11,13,15和16中为此目的描述的那些。对于基于本发明ctla4或cd80的mrna的表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:168,seqidno:169,seqidno:170,seqidno:171,seqidno:172和seqidno:173或seqidno:174,seqidno:175和/或seqidno:176。一种优选的方法实施方案包括使用基因ctla4和/或cd80的dna甲基化分析来预测对药物的反应,该药物适于改变特别是抑制ctla4和/或cd80的免疫调节作用。合适的药物例如是ipilimumab(mdx-010;商品名yervoy,bristol-myerssquibb),tremelimumab(ticilimumab,cp-675,206,pfizer,medimmune,astrazeneca)。
对于根据本发明的方法,更优选地是用于编码b7蛋白的其他免疫调节基因,基因是cd276,c10orf54,hhla2,ncr3lg1,cd86和vtcn1。基因cd276,c10orf54,hhla2,ncr3lg1,cd86和vtcn1编码本发明意义内的免疫检查点,其也属于免疫球蛋白超家族。基因cd276或cd276分子也被称为b7-h3,b7h3,4ig-b7-h3和b7rp-2。用于cd276的dna甲基化分析优选范围例如包括:转录编码区域(15:73683966-73714518,seqidno:181)以及启动子(15:73679515-73692168,seqidno:182和15:73693817-73698829,seqidno:184),cpg岛(15:73683810-73685004,seqidno:183),ctcf和转录因子结合位点(15:73699041-73706032,seqidno:185)和增强子(15:73674927-73679619,seqidno:186和15:73710442-73723236,seqidno:187)。用于cd276的甲基化分析的其他优选范围选自在实施例13,15,16和17中为此目的描述的那些。对于基于本发明cd276的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于序列:seqidno:241,seqidno:242,seqidno:243,seqidno:244,seqidno:245,seqidno:246,seqidno:247,seqidno:248,seqidno:249,seqidno:250,seqidno:251,seqidno:251和/或seqidno:253。一种优选的方法实施方案包括使用cd276基因的dna甲基化分析来预测对药物的反应,该药物适于改变特别是抑制cd276的免疫调节作用。合适的药物例如是enoblituzumab(mga271,mocrogenics)和mgd009(macrogenics)。
基因c10orf54或第10号染色体开放阅读段54也称为b7h5,gi24,pp2135,vista,b7-h5,dd1alpha和sisp1。用于c10orf54的dna甲基化分析的优选范围,例如是转录编码区域(10:71747559-71773498,seqidno:188),以及启动子(10:71759805-71784691,seqidno:189和10:71723534-71753287,seqidno:191)和/或ctcf结合位点(10:71738936-71750940,seqidno:190)。用于c10orf54的dna甲基化分析的进一步优选范围选自在实施例11,14和17中为此目的描述的那些。对于基于本发明c10orf54的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于序列:seqidno:192,seqidno:193和/或seqidno:194。一种优选的方法实施方案包括使用c10orf54基因的dna甲基化分析来预测对药物的反应,该药物适于改变特别是抑制c10orf54的免疫调节作用。合适的抑制剂例如是ca-170(curis)和jnj-61610588(janssenbiotech,inc.)。
基因hhla2或herv-hltr-关联蛋白2也被称为b7-h7,b7y,b7h7和b7-h5。用于hhla2的dna甲基化分析的优选范围例如是:转录编码区域(3:108296490-108378285),以及含有增强子、启动子和ctcf结合位点的调节区域(3:108291804-108313328,seqidno:36)和其他两个启动子(3:108310655-108313476,seqidno:195和3:108341108-108352706,seqidno:196)。用于hhla2的dna甲基化分析更优选的范围选自在实施例11,14,15和16中为此目的所描述的那些。对于基于本发明hhla2的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于区域:seqidno:197,seqidno:198,seqidno:199,seqidno:200,seqidno:201,seqidno:202,seqidno:203,seqidno:204,seqidno:205,seqidno:206和/或seqidno:207。一种优选的方法实施方案包括使用hhla2基因的dna甲基化分析来预测对药物的反应,该药物适于改变特别是抑制hhla2的免疫调节作用。
基因ncr3lg1或自然杀伤细胞的细胞毒性受体3配体1也被称为b7h6,dkfzp686o24166和b7-h6。ncr3lg1是免疫调节基因,其产物属于b7蛋白,并是本发明的免疫检查点。用于ncr3lg1的dna甲基化分析的优选范围例如是:转录编码区(11:17351726-17377341,seqidno:208),和启动子(11:17347524-17359073,seqidno:209)和/或cpg-岛(11:17351068-17354459,seqidno:210)。对于基于本发明ncr3lg1的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于区域:seqidno:254和/或seqidno:255。一种优选的方法实施方案包括使用ncr3lg1基因的dna甲基化分析来预测对药物的反应,该药物适于改变ncr3lg1和/或ncr3编码的受体的免疫调节作用。
基因cd86或cd86分子也称为b7-2,b70,lab72,b7.2和cd28lg2,这是已知的。由cd86编码的配体是b7-蛋白中的一种,并且是本发明的免疫检查点,其可以例如,结合由cd28和ctla4编码的受体。用于cd86的dna甲基化分析的优选范围例如包括:转录编码区域(3:122055366-122121139),启动子(3:122071817-122084231,seqidno:211;3:122053987-122061537,seqidno:212;3:122081939-122095873,seqidno:213;3:122096457-122110191,seqidno:214)和/或增强子(3:122119408-122125888,seqidno:215)。对于cd86的甲基化分析更优选的范围选自在实施例17中为此目的描述的那些。对于根据本发明的cd86的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:256,seqidno:257,seqidno:258,seqidno:259,seqidno:260,seqidno:261,seqidno:262,seqidno:263和/或seqidno:264。一种优选的方法实施方案包括使用cd86基因的dna甲基化分析来预测对药物的反应,该药物适于抑制cd86编码的配体和/或由ctla4和或cd28编码的受体的免疫调节作用。合适的药物例如是伊匹单抗(ipilimumab)(mdx-010;商品名称yervoy,bristol-myerssquibb),曲美母单抗(tremelimumab)(ticilimumab,cp-675,206,pfizer,medimmune,astrazeneca)。
基因vtcn1或含v-set结构域的t细胞活化抑制剂1也称为b7-h4,b7h4,b7s1,b7x,b7h.5,pro1291和vctn1,这是已知的。由vtcn1编码的蛋白质属于b7蛋白质,是根据本发明的免疫检查点。用于vtcn1的dna甲基化分析的优选范围包括:转录编码区域(1:117143587-117210960)和/或启动子(1:117193851-117226364)。对于根据本发明的cd86的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:377,seqidno:378和/或seqidno:379。一种优选的方法实施方案包括使用vtcn1基因的dna甲基化分析来预测对药物的反应,该药物适于抑制vtcn1编码的配体和/或其受体的免疫调节作用。
免疫调节基因kir2dl4,kir3dl1,kir3dl3,lag3和cd160编码为mhc:肽复合物结合共受体的蛋白。kir2dl4,kir3dl1,kir3dl3,lag3和cd160是本发明意义内的免疫检查点,属于免疫球蛋白超家族。kir基因kir2dl4,kir3dl1和kir3dl3编码杀伤细胞免疫球蛋白样受体(简称kir或kir受体)。基因kir2dl4或杀伤细胞免疫球蛋白样受体,有两个结构域,长胞质尾,4,例如也被称为kir103,kir103as,g9p,cd158d,15.212,kir-103as和103as。kir2dl4的dna甲基化分析的优选范围例如包括:转录编码区域(19:54803535-54814517,seqidno:216),启动子(19:54799627-54807083,seqidno:217;19:54791651-54800236,seqidno:229)和/或两个增强子(19:54803283-54804114,seqidno:218和19:54812823-54819942,seqidno:219)。kir2dl4的甲基化分析的其他优选范围选自在实施例11,14和17中为此目的描述的那些。对于根据本发明kir2dl4的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:220,seqidno:221,seqidno:222,seqidno:223,seqidno:224,seqidno:225,seqidno:226,seqidno:227和/或seqidno:228。一种优选的方法实施方案包括使用kir2dl4基因的dna甲基化分析来预测对药物的反应,该药物适于抑制kir2dl4编码的受体的免疫调节作用。合适的药物是例如lirilumab(bristol-myerssquibb)。
基因kir3dl1或杀伤细胞免疫球蛋白样受体,三个结构域,长胞质尾区,1也被称为nkb1,cd158e,cl-11,cl-2,cd158e2,nkat3,amb11,nkb1b,kir,kir3dl2,cd158e1,cd158e1/2,nkat-3,nkat3和kir3dl1/s1,这是已知的。kir3dl1的dna甲基化分析优选的范围例如包括:转录编码区域(19:54816468-54866993),和启动子(19:54808180-54831092,seqidno:230)和/或两个增强子(19:54812749-54821037,seqidno:231和19:54835785-54863422,seqidno:232)。kir3dl1的dna甲基化分析的其他优选范围选自在实施例11和14中为此目的描述的那些。对于根据本发明kir3dl1的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:233,seqidno:234,seqidno:235和/或seqidno:236。一种优选的方法实施方案包括使用kir3dl1基因的dna甲基化分析来预测对药物的反应,该药物适于抑制kir3dl1编码的受体的免疫调节作用,例如lirilumab(也称为iph2102/bms-986015,来自bristol-myerssquibb和innatepharma)和iph4102(innatepharma).。
基因kir3dl3或杀伤细胞免疫球蛋白样受体,三个结构域,长胞质尾区,3也被称为kir2ds2,cd158z,kir3dl7,kir44,cd158z和kirc1。对于kir3dl3的dna甲基化分析的优选范围包括(19:54724479-54736536,seqidno:237),以及启动子(19:54723156-54728908,seqidno:238)和/或两个增强子(19:54723473-54725230,seqidno:239和19:54735023-54744608,seqidno:240)。对于基于本发明kir3dl3的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:265。一种优选的方法实施方案包括使用kir3dl3基因的dna甲基化分析来预测对药物的反应,该药物适于抑制kir3dl3编码的受体的免疫调节作用,例如lirilumab(也称为iph2102/bms-986015,来自bristol-myerssquibb和innatepharma)。
基因lag3或淋巴细胞激活基因3例如也称为fdc和cd223。lag3的甲基化分析的优选范围例如为:转录编码区域(12:6772512-6778455,seqidno:161),和启动子(12:6770333-6774801,seqidno:349),具有ctcf结合位点的可变启动子(12:6777768-6781320,seqidno:350),以及增强子(12:6774685-6778062,seqidno:351)。lag3的dna甲基化分析进一步优选的范围是选自在实施例4,7,8,9和14中为此目的描述的那些。对于基于本发明lag3的mrna确定,例如优选检测以下序列或以下序列的一部分:seqidno:352,seqidno:353,seqidno:354和seqidno:355。一种优选的方法实施方案包括使用lag3基因的甲基化分析来预测对于抑制剂的反应,该抑制剂适于抑制lag3编码的受体的免疫调节作用,合适的抑制剂例如mgd013(macrogenics)和bms-986016(bristol-myerssquibb)。
基因cd160或cd160分子也被称为by55,nk28和nk1。cd160的dna甲基化分析的优选范围例如包括:转录编码区域(1:145719471-145739288,seqidno:361)以及启动子(1:145718367-145722818,seqidno:362),可变启动子(1:145735798-145741393,seqidno:364),ctcf-结合位点(1:145720204-145722255,seqidno:363)和/或两个增强子(1:145727546-145735069,seqidno:365和1:145708511-145719786,seqidno:366)。对于cd160的dna甲基化分析更优选的范围选自在实施例11,14和17中为此目的的描述的那些。对于基于本发明cd160的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:356,seqidno:357,seqidno:358,seqidno:359和/或seqidno:360。一种优选的方法实施方案包括使用cd160基因的dna甲基化分析来预测对药物的反应,该药物适于改变尤其是抑制由cd160编码的受体的免疫调节作用。
免疫调节基因tigit,btla,havcr2,btnl2和cd48编码蛋白tigit,btla,havcr2,btln2和cd48,它们属于免疫球蛋白超家族,并是本发明意义上的免疫检查点。
基因tigit或具有ig和itim结构域的t细胞免疫受体,例如也称为dkfzp667a205,vsig9,vstm3,flj39873和wucam,这是已知的。对于tigit的dna甲基化分析的优选范围例如包括:转录编码区域(3:114276913-114310288)和三个具有增强子和ctcf结合位点的调节区(3:114273873-114278448,seqidno:266;3:114306113-114321848,seqidno:267;3:114288458-114302904,seqidno:268)。tigit的dna甲基化分析的进一步优选范围选自在实施例4,7,8,11,14,15,16和17中为此目的描述的那些。对于基于本发明tigit的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:269,seqidno:270,seqidno:271,seqidno:272,seqidno:273,seqidno:274和/或seqidno:275。一种优选的方法实施方案包括使用tigit基因的dna甲基化分析来预测对药物的反应,该药物适于改变尤其是抑制tigit的免疫调节作用。
btla或b和t淋巴细胞相关基因也称为cd272和btla1。btla的dna甲基化分析的优选范围例如包括:转录编码区域(3:112463968-112499561)和启动子(3:112494210-112503447,seqidno:276),可选启动子(3:112460682-112487674,seqidno:279)和/或增强子(3:112501618-112513121,seqidno:277;3:112486680-112495554,seqidno:278;3:112451208-112463106,seqidno:280;3:112508297-112522457,seqidno:281)。用于btla的dna甲基化分析更优选的范围选自在实施例4,6,7,8,11,和14中为此目的描述的那些。对于基于本发明btla的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:282,seqidno:283,seqidno:284和/或seqidno:285。一种优选的方法实施方案包括使用btla基因的dna甲基化分析来预测对于药物的反应,该药物适于改变尤其是抑制btla的免疫调节作用。
基因havcr2或甲型肝炎病毒细胞受体2也被称为timd-3,cd366,tim-3,timd3,flj14428,tim3,havcr-2和kim-3。havcr2的dna甲基化分析的优选范围包括:转录编码区域(5:157085832-157142869)和启动子(5:157138356-157147783,seqidno:286),可变启动子(5:157097106-157121624,seqidno:287)和三个增强子(5:157144698-157161450,seqidno:288;5:157116666-157139926,seqidno:289和5:157080879-157096259,seqidno:290)。用于havcr2的dna甲基化分析更优选的范围选自在实施例11和14为此目的描述的那些。对于基于本发明havcr2的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:291,seqidno:292,seqidno:293,seqidno:294,seqidno:295和/或seqidno:296。一种优选的方法实施方案包括使用havcr2基因的dna甲基化分析来预测对于药物的反应,该药物适于改变,例如抑制havcr2的免疫调节作用。合适的抑制剂是抗tim-3抗体tsr-022(tesaro,inc)。
基因btnl2或丁酰胆碱2也称为btl-ii,btn7,hsblmhc1和ss2。用于btnl2的dna甲基化分析的优选范围包括:转录编码区域(6:32393963-32407128,seqidno:387),以及具有启动子和调节元件的区域(6:32387381-32413712,seqidno:387)。btnl2的dna甲基化分析的进一步优选范围选自在实施例17中为此目的描述的那些。对于基于本发明btnl2的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:389。一种优选的方法实施方案包括使用btnl2基因的dna甲基化分析来预测对于药物的反应,该药物适于改变btnl2的免疫调节作用。这尤其包括拮抗剂,其适于抑制由btnl2编码的免疫检查点。
基因cd48或cd48分子也称为bcm1,blast,blast1,mem-102,slamf2,hcd48和mcd48。cd48的dna甲基化分析的优选区域包括启动子和调节元件(1:160676386-160722581)和启动子(1:160703189-160716911,seqidno:390)。cd48的dna甲基化分析的其他优选范围选自在实施例17中为此目的描述的那些。对于基于本发明cd48的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:389。一种优选的方法实施方案包括使用cd48基因的dna甲基化分析来预测对于药物的反应,该药物适于改变cd48的免疫调节作用。
cd40,tnfrsf9,tnfrsf18,tnfrsf4和cd27是编码蛋白质cd40,tnfrsf9,tnfrsf18,tnfrsf4和cd27的免疫调节基因。cd40,tnfrsf9,tnfrsf18,tnfrsf4和cd27是肿瘤坏死因子受体超家族的受体,它们是本发明意义上的免疫检查点。
基因cd40或cd40分子,tnf受体超家族成员5也称为tnfrsf5,p50,bp50和cdw40。对于cd40的dna甲基化分析的优选范围包括:转录编码区域(20:46118272-46129863,seqidno:297),和启动子(20:46115599-46122305,seqidno:298),可变启动子(20:46106713-46115878,seqidno:299)和/或增强子(20:46121274-46143091,seqidno:300)。cd40的dna甲基化分析的进一步优选范围选自在实施例4,7,8,9和14中为此目的描述的那些。对于cd40的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:301,seqidno:302,seqidno:303,seqidno:304,seqidno:305,seqidno:306和/或seqidno:307。一种优选的实施方案包括使用cd40基因的dna甲基化分析来预测对于药物的反应,该药物适于改变cd40的免疫调节作用。这尤其包括激动剂,其能够激活由cd40编码的受体,例如cp-870,893(pfizer,vlst),dacetuzumab(seattlegenetics),chilob7/4(universityofsouthampton)(南安普敦大学)。特别地,这些药物可以选择那些能够抑制cd40的免疫调节作用的药物。合适的药物例如lucatumumab(novartis)。
基因tnfrsf9或肿瘤坏死因子受体超家族成员9也被称为4-1bb,cdw137,cd137和ila。对于tnfrsf9的dna甲基化分析的优选范围包括:编码转录物的区域(1:7915894-7943165,seqidno:308),和启动子(1:7937419-7950341,seqidno:309),具有ctcf结合位点,增强子和可变启动子的调控元件(1:7910016-7923298,seqidno:312),增强子(1:7922095-7939183,seqidno:310)和/或cpg岛(1:7949284-7956816,seqidno:311)。对于tnfrsf9的dna甲基化分析进一步优选的范围选自在实施例4,7,8,11,14,15,.16和17中为此目的描述的那些。对于基于本发明的tnfrsf9的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:313,seqidno:314,seqidno:315和/或seqidno:316。优选的方法实施方案包括使用tnfrsf9基因的dna甲基化分析来预测对于药物的反应,该药物适于改变tnfrsf9的免疫调节作用。这尤其包括激动剂,其能够激活由tnfrsf9编码的受体,合适的药物例如是pf-05082566(pf-566,pfizer)和urelumab(bms-663513,bristol-myerssquibb)。
基因tnfrsf25或肿瘤坏死因子受体超家族成员25也被称为wsl-lr,wsl,apo3,tnfrsf12,tr3,lard,ddr3,tramp,wsl-1,dr3,wsl1和apo-3。对于tnfrsf25的dna甲基化分析的优选范围包括:编码转录物的区域(1:6461151-6466195,seqidno:317),以及可变启动子(1:6458470-6463974,seqidno:318),两个cpg岛(1:6459713-6463353,seqidno:319;1:6465602-6466846,seqidno:320),和另外的启动子(1:6464008-6468536,seqidno:321)和/或增强子(1:6468908-6478190,seqidno:322)。对tnfrsf25的dna甲基化分析更优选的范围选自在实施例14和17为此目的描述的那些。对于基于本发明的tnfrsf25的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:323,seqidno:324,seqidno:325,seqidno:326,seqidno:327和/或seqidno:328。优选的方法实施方案包括使用tnfrsf25基因的dna甲基化分析来预测对于药物的反应,该药物适于改变tnfrsf25的免疫调节作用。这尤其包括激动剂,其适于激活由tnfrsf25编码的受体。
基因tnfrsf18或肿瘤坏死因子受体超家族成员18也称为gitr,aitr,cd357和gitr-d。对于tnfrsf18的dna甲基化分析的优选范围包括:编码转录物的区域(1:1203508-1206691,seqidno:329)和/或启动子(1:1195867-1210392,seqidno:330)。对于基于本发明的tnfrsf18的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:331,seqidno:332,seqidno:333和/或seqidno:334。对于tnfrsf18的甲基化分析更优选的范围选自在实施例17中为此描述的那些。一种优选的方法实施方案包括使用tnfrsf18基因的dna甲基化分析来预测对于药物的反应,该药物适于改变tnfrsf18的免疫调节作用。这尤其包括激动剂,其适于激活由tnfrsf18编码的受体,例如trx518(gitrinc.)。
基因tnfrsf4或肿瘤坏死因子受体超家族成员4也被称为imd16,ox40,txgp1l,cd134和act35。对于tnfrsf4的dna甲基化分析的优选范围包括:编码转录物的区域(1:1211326-1214138,seqidno:335),启动子(1:1209894-1215938,seqidno:336)和/或cpg岛(1:1213505-1214288,seqidno:337)。对于根据本发明的tnfrsf4的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:338,seqidno:339和/或seqidno:340。tnfrsf4的甲基化分析的进一步优选范围选自在实施例17中为此目的描述的那些。一种优选的方法实施方案包括使用tnfrsf4基因的dna甲基化分析来预测对于药物的反应,该药物适于改变tnfrsf4的免疫调节作用。这尤其包括激动剂,其适于激活由tnfrsf4编码的受体,例如medi6469(9b12,medimmunellc,astrazeneca,agonox),hu106-22和hu119-122(utmdacc)。
基因cd27或cd27分子也被称为s152,t14,tnfrsf7,s152,lpfs2或tp55。对于cd27的dna甲基化分析的优选范围包括:编码转录物的区域(12:6444867-6451718,seqidno:341),两个启动子(12:6442585-6448021,seqidno:342和12:6448982-6453895,seqidno:343)和/或三个增强子(12:6453736-6456010,seqidno:344;12:6447474-6449430,seqidno:345和12:6439173-6442795,seqidno:346)。cd27的dna甲基化分析的其他优选范围选自在实施例6和17中为此目的描述的那些。对于根据本发明的cd27的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:347和/或seqidno:348。一种优选的方法实施方案包括使用cd27基因的dna甲基化分析来预测对于药物的反应,该药物适于改变cd27的免疫调节作用。这尤其包括激动剂,其适于激活由cd27编码的受体,例如varlilumab(celldextherapeutics)。
adora2a基因是编码腺苷2a受体的免疫调节基因。腺苷2a受体是根据本发明意义上的免疫检查点,其通过与代谢物腺苷的结合发挥免疫调节作用。基因adora2a或腺苷a2a受体也称为a2ar,adora2或rdc8。对于adora2a的dna甲基化分析的优选范围包括:编码转录物的区域(22:24417879-24442360,seqidno:380),具有cpg岛的启动子(22:24422358-24434469,seqidno:381),增强子(22:24418577-24423493,seqidno:382)和编码adora2a及其反义rna的区域(22:24420336-24501503)。adora2a的dna甲基化分析的其他优选范围选自在实施例17中为此目的描述的那些。对于根据本发明的adora2a的mrna表达分析,适于检测至少以下转录物的序列的一部分,其cdna至少部分地对应于:seqidno:383,seqidno:384,seqidno:385,和/或seqidno:386。一种优选的方法实施方案包括使用adora2a基因的dna甲基化分析来预测对于药物的反应,该药物适于改变腺苷2a受体的免疫调节作用。这尤其包括拮抗剂,其适于抑制由adora2a编码的受体,例如pbf-509,istradefylline,st1535,st4206,tozadenant,v81444,cpi-444,preladenant,vipadenant,sch58261,atl801。
可替代地,本发明免疫调节基因的dna的甲基化分析可以结合其它预后性和/或预测性生物标志物进行。特别适合于此的是braf和egfr基因的突变,通过它们可以预测对于braf和egfr的抑制剂的反应。也可以将免疫调节基因的甲基化分析与趋化因子和趋化因子受体的甲基化分析和/或突变分析结合使用,例如趋化因子家族cxc和cx3c。例如,这可以用于预测对ulocuplumab疗法的反应。根据本发明的免疫调节基因的甲基化分析也可以与基因il2rb,cxcl12,cxcr4和cxcr7的甲基化分析相结合。
原则上,对预后的评估也可以在用至少一种药物对病人进行免疫疗法治疗之前,期间或之后进行,其中该药物用于改变免疫检查点的免疫调节作用。预测也可包括患者的恶性疾病对使用至少一种这样的药物的免疫治疗的反应。免疫调节作用的作用可以通过以下方式来实现,即药物与所分析的免疫检查点本身或其受体或配体相互作用。例如,免疫检查点可以包括配体cd274和/或pdcd1lg2,因此药物通过与相应的受体pdcd1相互作用来改变,尤其是抑制cd274和/或pdcd1lg2的免疫调节作用,例如其中药物阻断cd274或pdcd1lg2到pdcd1受体的结合。特别地,所谓的免疫检查点抑制剂可以考虑作为药物,其能够降低或防止共抑制性或抗炎性免疫检查点的免疫调节作用。这些免疫检查点抑制剂的实例是nivolumab,ipilimumab,帕母单抗和其他在上面已经提到的药物。另一种可能是可以激活根据本发明的免疫检查点的免疫调节作用的药物。合适的活化剂包括上面已经提到的那些。也可以根据基于本发明的各种不同免疫调节基因的dna甲基化分析来从药物中选择那些在免疫疗法中有良好反应性的药物。这样的组合疗法往往特别有效,但由于涉及高成本需要非常慎重的考虑,因此,要求特别可靠的对各类药物的反应的预测。这是本发明的方法所首次提供的。
根据对患有恶性疾病的患者选择个性化的药物以提供免疫疗法治疗的方法,这里接着描述本发明的第二方面。本方法的特征还在于,对恶性肿瘤细胞和/或与恶性疾病细胞相互作用的t淋巴细胞的至少一个基因进行dna甲基化分析,其中免疫检查点的免疫调节基因从以下成分中选择:b7蛋白及其受体(即结合b7蛋白的受体),mhc:肽复合物结合共同受体,肿瘤坏死因子受体超家族的成员tnfrsf9,cd40,tnfrsf4,tnfrsf18,和cd27,免疫球蛋白超家族tigit,btla,havcr2,btnl2和cd48和腺苷结合腺苷2a受体的成员,并且基于dna甲基化分析的结果选择药物。
还可以选择药物使得它可以改变免疫检查点的免疫调节作用,例如,作为免疫检查点抑制剂,能够降低或防止共抑制性或抗炎性免疫检查点的免疫调节作用;或者作为免疫检查点激活剂,能够增强共刺激或促炎性免疫检查点的免疫调节作用。
特别是,当dna甲基化分析指示在恶性疾病和/或t淋巴细胞的细胞中相应的免疫检查点表达时,选择药物,因为免疫检查点的表达提供了使用这种药物进行免疫疗法治疗的攻击点,因此对该种免疫疗法的反应也很可能是充分的。
本发明的第三方面因此涉及使用患者恶性疾病细胞和/或与恶性疾病细胞相互作用的t淋巴细胞的至少一种免疫调节基因的dna甲基化分析来确定预后、预测和/或个性化选择药物以对病人进行免疫疗法治疗。同样地,免疫调节基因编码选自以下的免疫检查点:b7蛋白及其受体(即结合b7蛋白的受体),mhc:肽复合物结合共同受体,肿瘤坏死因子受体超家族的成员tnfrsf9,cd40,tnfrsf4,tnfrsf18和cd27,免疫球蛋白超家族tigit,btla,havcr2,btnl2和cd48和腺苷结合的腺苷2a受体的成员。
本发明的第四方面涉及将恶性疾病细胞和/或与恶性疾病细胞相互作用的t淋巴细胞的一种免疫调节基因中至少一个cpg二核苷酸的甲基化的存在、不存在或程度用作生物标记物来个性化选择药物,以对病人进行免疫治疗,其中免疫调节基因编码选自以下的免疫检查点:b7蛋白及其受体(即结合b7蛋白的受体),mhc:肽复合物结合共同受体,肿瘤坏死因子受体超家族的成员tnfrsf9,cd40,tnfrsf4,tnfrsf18和cd27,免疫球蛋白超家族tigit,btla,havcr2,btnl2和cd48和腺苷结合的腺苷2a受体的成员。
另外,只要适用,第二方面,第三方面和第四方面的其他特征和优选实施方案对应于第一方面的那些。
根据本发明的第五方面,提供一种用于实施根据第一或第二方面的方法或用于第三方面或第四方面的用途的试剂盒。该试剂盒包括至少一种用于dna甲基化分析的寡核苷酸对,其中在dna中含有的胞嘧啶转化为尿嘧啶或具有与胞嘧啶可区分的碱基配对体和/或分子量的另一种碱基后,所述寡核苷酸对适于与恶性疾病细胞和/或t淋巴细胞的dna中的免疫调节基因序列杂交,以扩增和/或检测该序列。
所述试剂盒还可包含一种或多种寡核苷酸对,所述寡核苷酸对被设计为与经转换的dna中的免疫调节基因自身或其他免疫调节基因的其他序列杂交,来对其他序列进行dna甲基化分析,以扩增和/或检测该序列。
所述试剂盒还可包含一种寡核苷酸对用于对mrna表达分析,其中所述寡核苷酸对被设计为与来自恶性疾病细胞和/或t淋巴细胞中的免疫调节基因的至少一种转录变体的mrna或由该mrna生成的cdna的序列杂交,以扩增和/或检测该序列。
使用寡核苷酸对来扩增和/或检测的免疫调节基因的优选区域和序列与第一方面的那些相似。
该试剂盒优选包括用于实施根据第一和/或第二方面的方法和/或用于根据第三或第四方面的用途的说明书。
下面将参考示例性实施方案和实验结果更详细地描述本发明。这些实施例是说明性的,并不限于特定细节。
实施例1:基于免疫调节基因pdcd1的dna甲基化分析,确定根治性切除前列腺癌术后患者的预后
根据本发明的方法实施例,患者预后的确定可以在局部受限的(局部的,未扩散的)前列腺癌中进行,例如在已经进行了前列腺根治性切除和位于前列腺中的肿瘤的根治性切除后。
首先,从肿瘤中获取dna。例如,可以使用蛋白酶k裂解肿瘤组织,随后用二氧化硅离心柱提取dna。为了从肿瘤获得dna,可商购市售的试剂盒,例如qiaampdnaminikit(qiagenn.v.,helden,德国)。随后通过亚硫酸氢盐转化提取的dna,使得基本上所有胞嘧啶脱氨基为尿嘧啶,而甲基化的胞嘧啶则保持不变。随后进行甲基化分析,其中例如使用infiniumhumanmethylation450beadchip芯片(illuminainc.,sandiego,ca,usa),其适于根据制造商的说明使用。humanmethylation450beadchip原始数据的生成如tcga研究网络(http://cancergenome.nih.gov/)中所述。总共生成并回顾性分析了具有已知预后的417名前列腺癌患者的肿瘤的原始数据。
对于本发明基因pdcd1的dna甲基化分析,首先基于humanmethylation450beadchip原始数据计算得到相对甲基化值。为此,使用了pdcd1基因中的cpg二核苷酸,其由infiniumhumanmethylation450beadchip中的珠对(bead-paare)cg00795812,cg27051683,cg03889044,cg17322655和cg20805133覆盖。这些珠对与经亚硫酸氢盐转化的dna结合,所述dna在转化之前具有序列seqidno:20,seqidno:21,seqidno:22,seqidno:23和seqidno:24。首先,根据每个所述珠对和每个患者样品的所述原始数据计算甲基化值。这是通过将结合甲基化变体(s_m)的珠对的信号与结合未甲基化的dna(s_u)珠对的信号相比较而实现的。珠包含结合的寡核苷酸,并且在本文中也称为探针。基于比率的形成计算的dna甲基化根据如下公式进行:甲基化=(探针强度s_m)/((探针强度s_m)+(探针强度s_u))。随后,对如此测定的具有序列seqidno:20,seqidno:21,seqidno:22,seqidno:23和seqidno:24的五个基因座中的每一个的甲基化值进行算术取平均值。然后,根据他们已知的预后,将患者按照如此测定的的甲基化值分成两组,由此得出pdcd1基因座的甲基化阈值为57.5%。pdcd1基因座的甲基化低于阈值57.5%的组,在下文中称为低pdcd1甲基化的组。相应地,pdcd1基因位点的甲基化高于阈值57.5%的组,在下文中称为高pdcd1甲基化的组。
图1显示了通过使用本发明的方法,将患者分为两组,其中低pdcd1甲基化的患者组比高pdcd1甲基化的患者组具有显著更好的预后,这里预后定义为生化复发的发生-也就是前列腺癌特异性血液参数psa的升高-的预计时间。低pdcd1甲基化的组的psa无复发生存期平均为92个月,而高pdcd1甲基化的组平均为79个月,显示出其psa无复发生存期短了13个月。因此,应用根据本发明的方法使得可以将患者分成用于区分psa复发的不同风险组。
因此,可以确定刚刚切除肿瘤并且不知晓其未来疾病进程的患者的预后。为此目的,可以例如通过infiniumhumanmethylation450beadchip测定该患者肿瘤中pdcd1基因的dna甲基化。例如,如果该甲基化低于57.5%的阈值,那么该患者可以被分类到具有更好预后的、平均具有92个月的无复发存活的患者组。例如,如果患者肿瘤中pdcd1基因的甲基化高于阈值,那么该患者可能更早发生psa复发。然后该患者可以从额外的辅助治疗中受益。例如,还可以在随访期间密切监测这些患者的进展,从而也可以在早期阶段诊断psa复发的发生,从而可以开始合适的治疗措施。
实施例2:基于免疫调节基因cd274的dna甲基化分析确定根治性切除术后前列腺癌患者的预后
通过实施根据本发明的方法,还能够通过cd274基因内的cpg二核苷酸的dna甲基化分析确定恶性疾病患者的预后。例如,在患者接受完全去除(根治性切除)前列腺和位于前列腺中的肿瘤后,基于肿瘤内cd274基因的甲基化获得局部受限的前列腺癌症患者的预后。
总共回顾性分析了具有已知预后的417名患者的肿瘤样本。dna甲基化分析如实施例1中所述进行。为了基于infiniumhumanmethylation450beadchip原始数据计算cd274基因位点的相对甲基化值,使用了五个珠对cg15837913,cg02823866,cg14305799,cg13474877和cg19724470,它们使得能够进行基因组中序列seqidno:8,seqidno:9,seqidno:10,seqidno:11和seqidno:12的cpg二核苷酸甲基化分析。如实施例1中描述的pdcd1基因的分析,对每个患者的五个珠对中的每一个计算相对的甲基化值,这是如实施例1中所描述的通过将一个柱对中特定珠的未甲基化水平和甲基化水平彼此相对比而实现的。随后,将这五个甲基化值平均以得到每个患者样品的相对甲基化值。然后根据各自已知的预后,基于如此测定的患者的甲基化值将患者划分为两组,由此得出cd274基因位点的相对甲基化阈值为52.75%。cd274基因位点的甲基化低于52.75%的阈值的组具有337名患者,在下文中称为低cd274甲基化的组。相应地,cd274基因位点的甲基化高于52.75%的阈值的组,在下文中称为高cd274甲基化的组。该组包括80名患者。
图2a显示了通过使用本发明的方法将患者分为两组,低cd274甲基化的患者组比高cd274甲基化的患者组具有显著更好的预后,在本实施例中预后定义为生化复发的发生-也就是前列腺癌特异性血液参数psa的升高-的预计时间。低cd274甲基化的组的psa无复发生存期平均为2822天,而高pdcd1甲基化的组平均为1578天,显示出其psa无复发生存期短了1244天。因此,应用根据本发明的方法使得可以将患者分成用于区分psa复发的不同风险组。
还可以通过实时pcr进行dna甲基化分析,以根据本发明确定恶性疾病患者的预后。在本实施例中,回顾性分析了259名患有前列腺肿瘤并具有已知预后的患者,这是与先前描述的417名患者不同的患者组。
为了借助实时pcr进行dna甲基化分析,制备厚度为10μm的组织薄切片并将其安装在载玻片上。基于he切片,通过病理学检查鉴定肿瘤区域。然后使用手术刀将这些肿瘤区域从玻璃物体上刮下。根据制造商的说明,用innuconvertbisulfitall-in-onekit(analytikjena,jena,德国)获得肿瘤区域的经亚硫酸氢盐转化的dna。随后,使用nanodropnd-1000分光光度计(thermofisherscientific,waltham,ma,美国)定量经转化的dna的量。通过使用定量实时pcr(qpcr)以甲基化特异性方式扩增并同时定量cd274基因位点来进行前列腺肿瘤的dna甲基化分析。在这种情况下,使用双链或多链pcr,其中除了确定相同反应中的甲基化外,还测定总dna。总dna的测定可以例如通过使用其靶序列不含cpg二核苷酸并由此其靶序列独立于甲基化而扩增的引物和探针来进行。在本例中,为了测定总dna,扩增了actb基因中的基因位点。基因组中的该基因座具有seqidno:32的序列,并且在通过亚硫酸氢盐转化后具有seqidno:7的序列。该序列使用具有序列seqidno:4和seqidno:5的引物进行扩增。扩增之后用序列seqidno:6的探针进行序列特异性检测,其在5’端加上荧光染料atto647n,在3’端加上猝灭剂bhq-2。随后用具有seqidno:13和seqidno:14的引物进行cd274基因位点的甲基化特异性扩增。这些引物扩增了由序列seqidno:33经过亚硫酸盐转化产生的序列。在完全甲基化的情况下,基因组中的该转化区域具有序列seqidno:16。用序列seqidno:15的探针进行甲基化特异性检测,其在5’端加上荧光染料6-fam,在3’端加上猝灭剂bhq-1。前列腺肿瘤的经转化的dna的甲基化状态是通过deltadelta-ct的方法来计算,并用相对于具有100%甲基化的标准dna的百分比来表示。使用的标准dna是人为甲基化的dna(cpgenometmuniversalmethylateddna;merckmillipore,darmstadt,德国),其先前使用innuconvertbisulfiteall-in-onekit(试剂盒)根据制造商的说明书进行转化。在本实施例中,cd274的甲基化在20μl的pcr反应液中的实时pcr一式三份地独立测量,其特征在于,反应液适于包括以下组成:35mmtris-hcl,ph8.4;6mmmgcl2;50mmkcl,4%甘油,每种dntp(dttp,datp,dgtp,dctp)0.25mm,2ufaststarttaqdna聚合物(rocheappliedscience,penzberg,德国),每种引物0.4μm和每种检测探针0.2μm。使用ab7500快速实时pcr系统(lifetechnologiescorporation,carlsbad,ca,usa)进行qpcr。合适的温度曲线例如包括以下步骤:在95℃下20分钟,随后在56℃下45秒,在95℃下15秒;循环45次。
图2b显示了通过使用本发明的方法使用实时pcr确定cd274基因座的甲基化,并确定前列腺癌患者的预后。259名患者可以基于dna甲基化分析分为具有不同的预后的两组。患者肿瘤的甲基化超过0.975%,显示出具有平均93个月的psa无复发生存期,而患者肿瘤的甲基化低于0.975%,显示出平均延长了21个月的psa无复发生存期(114个月)。
例如,可以确定与上述实时pcr结果类似而预后未知的患者的肿瘤样品中cd274基因座的甲基化。例如,如果该甲基化高于0.975%,则患者可能具有不利的预后,可能平均93个月的非复发存活期。相反,如果肿瘤中的甲基化低于该阈值,则患者可能具有更有利的预后,平均114个月的非复发存活期。
在该实施例中,使用两种不同的技术,即实时pcr和infiniumhumanmethylation450beadchip执行本发明的方法。基于甲基化的阈值,其为52.75%(infiniumhumanmethylation450beadchip)和0.975%(实时pcr),将患者分为具有良好预后的组和具有不良预后的组。对于本领域技术人员显而易见的是,可以根据用于dna甲基化分析的方法确定用于预后组分类的适当阈值。另一种可能是不同的方法的相互校准,以便可以直接比较各dna甲基化分析的结果。例如,可以通过不同方法进行具有确定的甲基化百分比的一种或多种标准dna样品的dna甲基化分析的校准。通过为每种方法提供待测免疫调节基因的dna甲基化分析结果,以及相关的标准dna样品的dna甲基化分析结果,可以校准测量结果,使得来自不同方法的经校准的测量结果具有可比性。
实施例3:基于免疫调节基因pdcd1,pdcd1lg2和cd274的dna甲基化分析确定恶性黑色素瘤患者的预后
该实施例显示了根据本发明确定470名患有恶性黑素瘤的患者的预后。如实施例1中所述,使用infiniumhumanmethylation450beadchip产生dna甲基化分析的结果。为了确定相应基因的相对甲基化值,首先如实施例1中所述对infiniumhumanmethylation450beadchip的每个选择的珠测定相对甲基化值。然后对每个基因多达六个珠对的的相对甲基化值取平均值,以获得特定基因的代表性甲基化值。为了对cd274进行甲基化分析,使用珠对cg15837913(seqidno:8),cg02823866(seqidno:9),cg14305799(seqidno:10),cg13474877(seqidno:11)和cg19724470(seqidno:12)。这些珠例如允许确定cd274基因中序列区域seqidno:1,seqidno:33和seqidno:76中的cpg二核苷酸的甲基化。为了对pdcd1进行甲基化分析,使用珠对cg00795812(seqidno:20),cg03889044(seqidno:22),cg17322655(seqidno:23),cg20805133(seqidno:24)和cg27051683(seqidno:369)。这些珠例如允许确定pdcd1基因中序列区域seqidno:3和seqidno:29中的cpg二核苷酸的甲基化。为了对pdcd1lg2进行甲基化分析,使用珠对cg07211259(seqidno:31)。这些珠例如允许确定pdcd1lg2基因中序列区域seqidno:84中的cpg二核苷酸的甲基化。
图3显示了基于dna甲基化分析将470名恶性黑素瘤患者回顾性划分成几个预后组。其显示了根据其预后基于肿瘤中基因pdcd1(图3a),cd274(图3b)和pdcd1lg2(图3c)的甲基化分组的患者的kaplan-meier存活分析。组i包括其肿瘤在相应基因座处具有低于阈值的低甲基化的患者。组ii包含其肿瘤的甲基化超过相应基因阈值的患者。例如,将以下甲基化水平设定为患者分组的阈值:pdcd1lg2为12.84%,cd274为13.11%,pdcd1为20.54%。对于所分析的全部三种基因,dna甲基化分析显示肿瘤中甲基化升高的患者表现出不太有利的疾病进程。因此,使用本发明的方法使得可以基于免疫检查点基因pdcd1和编码相关配体的基因cd274的dna甲基化来确定恶性黑素瘤患者的预后组。此外,结果表明,根据本发明,编码免疫检查点pdcd1的配体的另一种基因可与pdcd1lg2一起用于确定预后。
该实施例表明本发明的方法不仅局限于如实施例1和实施例2中所述的一种恶性疾病,前列腺癌,而且在其他恶性疾病中,例如为恶性黑素瘤确定预后也是可能的。因此,基于这些基因的dna甲基化分析确定预后不限于单个实体,而是也可以应用到其他恶性肿瘤。因此,本发明解决了对传统预测方法中各个实体的适用性有限的问题。
此外,在该实施例中可以显示,通过根据本发明的方法实施方案,可以确定将来各种健康状况的预后。在实施例1和实施例2中,可以正确预测psa复发的发生。psa复发是与前列腺癌相关的性质。而在前述实施例3中,可以预测患者的死亡。
实施例4:基于免疫调节基因tigit,tnfrsf9,lag3,btla,ctla4和cd40的dna甲基化分析确定恶性黑色素瘤患者的预后
根据本发明的方法还可以基于免疫检查点基因tigit,tnfrsf9,lag3,btla,ctla4和cd40的甲基化来评估患有恶性黑素瘤的患者的预后。在本实施例中,如实施例1所述,使用infiniumhumanmationation450beadchip(illumina,inc.,sandiego,ca,usa)的dna甲基化分析结果确定基因的相对甲基化。随后,确定基因tigit,tnfrsf9,lag3,btla,ctla4和cd40的相对甲基化。为此目的,如实施例1中所述,形成珠对的特定珠的甲基化和未甲基化状态的比率。对于基因btla,使用了珠对cg24157392和cg19281794。这些珠结合人类dna中经亚硫酸氢盐转化之前的序列seqidno:37或seqidno:37,因此允许例如确定序列区域seqidno:276的甲基化。两个珠对各自由一对珠组成,其中一个珠在亚硫酸氢盐转化之前与未甲基化序列结合,另一个珠在转化前与甲基化序列结合。用这两个珠对形成比值,例如对于珠对cg24157392以下面方式示出:甲基化=(cg24157392_m强度)/((cg24157392_m强度)+(cg24157392_u强度)),其中所述cg24157392_m是结合甲基化变体的珠,cg24157392_u是结合未甲基化变体的珠。对于珠对cg19281794使用相同的方法。然后计算btla的相对甲基化值,其中相对甲基化根据由珠对对cg24157392和cg19281794算出。
因此,确定了基因tigit,tnfrsf9,lag3,btla,ctla4和cd40的相对甲基化。为了确定tnfrsf9的相对甲基化,使用了珠对cg14614416(seqidno:51),cg14153654(seqidno:54),cg17123655(seqidno:55),cg18025409(seqidno:57),cg06956444(seqidno:58)和cg08840010(seqidno:59),其中,例如可以确定基因组中序列区域seqidno:308,seqidno:309和seqidno:321的甲基化。为了确定cd40的甲基化,使用了珠对cg19785066(seqidno:47),cg09053081(seqidno:48)和cg21601405(seqidno:49),其例如适于确定序列区域seqidno:297和seqidno:298的甲基化。为了确定tigit的甲基化,使用了珠对cg02771886(seqidno:61),cg08723913(seqidno:62),cg17164827(seqidno:63),cg04885775(seqidno:64),cg23637607(seqidno:65)和cg16358924(seqidno:66),其例如适于确定序列区域seqidno:267的甲基化。为了确定基因ctla4序列区域seqidno:162的甲基化,使用了珠对cg08460026(seqidno:39)。为了确定lag3的相对甲基化,使用了珠对cg04153135(seqidno:40),cg20652042(seqidno:41)和cg01820374(seqidno:42),其例如适于确定序列区域seqidno:161,seqidno:349和seqidno:351的甲基化。
图4显示了基于本发明的dna甲基化分析将470名恶性黑素瘤患者回顾性划分成几个预后组。其显示了基于肿瘤中基因tigit(图4a),tnfrsf9(图4b),lag3(图4c),btla(图4d),ctla4(图4e)和cd40(图4f)的甲基化而分组的患者的kaplan-meier存活分析,组i包括其肿瘤在相应基因座处具有低于阈值的低甲基化的患者。组ii包含那些肿瘤甲基化超过相应基因阈值的患者。例如,将以下甲基化水平设定为患者分组的阈值:tigit为86.41%,tnfrsf9为57.80%,lag3为66.94%,btla为91.31%,ctla4为13.10%,cd40为17.16%。对于所有这些分析的基因,肿瘤中甲基化升高到高于阈值的患者比肿瘤中甲基化低于阈值的患者,具有显著更加不利的疾病进程。
实施例5:基于免疫调节基因pdcd1的dna甲基化分析确定头颈部区域鳞状细胞癌患者的预后
图5显示了通过根据本发明的方法对528名患有头颈部区域的鳞状细胞癌的患者的预后组的回顾性划分。临床终点定义为死亡。在来自肿瘤组织的dna中测量免疫检查点基因pdcd1的dna甲基化分析。高pdcd1甲基化的患者表现出比低pdcd1基因位点甲基化的患者更差的结果。患者的分组是使用中间值作为阈值分为两组,也就是说,该阈值被选择为使得一半(n=264名患者)的患者样品的甲基化水平高于所述阈值,而另一半(n=264名患者)低于所述阈值。
也可以通过实时pcr进行pdcd1的dna甲基化分析。用于实时pcr的合适条件和组合物类似于实施例2。下列寡核苷酸被用于pcr:seqidno:25(正向引物),seqidno:26(反向引物)和seqidno:27的检测探针。用fam和bhq-1标记检测探针。使用的引物允许扩增序列seqidno:368的经亚硫酸氢盐转化的dna。该序列在亚硫酸氢盐t转化之前具有序列seqidno:367。对120例头部颈部鳞状细胞癌患者的肿瘤样本进行回顾性分析。根据其预后,将患者基于dna甲基化分析分组,一组具有40例低甲基化的患者,另一组具有80例高甲基化的患者,具有高甲基化的患者组比低甲基化的患者组总体存活时间显著更少(p值<0.05)。
在实施例1已表明,根据本发明的方法可以基于基因pdcd1的甲基化确定前列腺癌患者的预后。这些前列腺癌是由前列腺的腺上皮产生的癌。因此这些癌属于腺癌。在实施例3中,还可以显示根据本发明的方法使得恶性黑素瘤患者的预后成为可能。黑色素瘤不属于癌,因为它们不是来自上皮细胞,而是来自黑素细胞。因此,与癌相比,黑色素瘤代表了根本不同的恶性疾病。在本实施例5中,还显示本发明能够确定患者头颈部区域鳞状细胞癌的预后。这些鳞状细胞癌,如同腺癌,属于癌,即它们从上皮细胞中出现。与腺癌相反,鳞状上皮不从腺体出来,而是来自鳞状上皮。因此可以证明,根据本发明的方法不仅可以确定不同来源的癌症的预后,还可以确定非癌症的恶性疾病的预后。这些结果进一步证明本发明的方法对于确定预后和/或预测不同恶性疾病具有普遍适用性。
实施例6:通过dna甲基化分析确定免疫检查点基因的mrna表达
根据本发明的方法还可以确定免疫调节基因编码的免疫检查点的表达,例如如下所示基于基因btla和cd27的mrna表达。
mrna的确定例如提取mrna之后,准备总rna的cdna文库,并最终基于下一代测序分析来确定。例如,rna可以借助rneasyminikit(qiagen,hilden)按照制造商的说明书从肿瘤组织中提取。随后,例如,使用truseqrnalibrarypreparationkitv2(illumina)按照相应的说明书来制备文库,并通过hiseqpe(paired-end)clusterkitv4cbot(illumina)在hiseq2500下一代测序仪(nextgenerationsequencer)(illumina)上进行分析。将mrna的测量结果或特定序列的mrna分子的量与总mrna分子量进行对比如tcga研究网络(http://cancergenome.nih.gov/)中所述。
图6显示了470名恶性黑色素瘤患者组织中的dna甲基化与mrna表达的相关性,其中图6a代表btla,图6c代表cd27。可以看出,所有的相应基因座的甲基化小于50%的肿瘤,其mrna的表达超过10(btla,图6a)或超过100(cd27,图6c)。图6b显示,黑色素瘤肿瘤的btlamrna表达>10的患者比肿瘤具有较低mrna表达的患者具有显著更好的预后。类似于btla,在图6d中显示,肿瘤的cd27mrna表达>100的黑色素瘤患者具有比其肿瘤具有较低mrna表达的患者显著更好的预后。
基因btla和cd27的表达不仅可以用于病人预后的确定,而且还指示对治疗的可能反应,例如当治疗被引向对应的基因治疗。该实施例表明,基于肿瘤中基因座的甲基化可以确定mrna表达,并且可以用于确定患者的预后。这解决了由于mrna的低稳定性而直接测定mrna本身在临床常规中受限的问题。此外,该实施例显示基于dna甲基化分析确定患者的预后,也可同时估计所分析的免疫调节基因的表达。因此患者可以有资格接受很可能起作用或很可能不起作用的治疗,这取决于基因产物即免疫检查点的表达。特别适用于此目的的是直接针对免疫检查点或其相互作用配偶体的治疗。特别是,针对这些基因产物的单克隆抗体的免疫疗法适用于此目的。
实施例7:使用dna甲基化分析和mrna表达分析的组合确定恶性黑素瘤患者的预后
在470名患有恶性黑色素瘤并具有已知预后的患者的肿瘤中进行cd274基因的dna甲基化分析和mrna表达分析。如实施例1,3和6中所述进行mrna表达和dna甲基化的测定。
图7a显示基因cd274的具有各自阈值的mrna表达和dna甲基化的散点图,基于该阈值,回顾性划分具有高和低mrna表达或dna甲基化的组。图7a显示显著负相关(spearman’sρ=-0.546,p<0.001)的dna甲基化和mrna表达。根据本发明,基于引入的甲基化阈值13.11%,患者可以分为具有较高(组ii+iii)和较低(组i+iv)dna甲基化的组。类似地,在图7a中引入mrna表达的垂直阈值18.23,该图分为具有高(组iii+iv)和低(组i+ii)mrna表达的两组。图7b显示肿瘤中具有cd274高甲基化(组ii+iii)与低甲基化(组i+iv)的患者总生存期的kaplan-meier分析对比。与i+iv组的患者相比,ii+iii组患者预后较差。图7c显示了肿瘤中具有cd274高mrna表达(组iii+iv)与低甲基化表达(组i+ii)的患者总生存期的kaplan-meier分析对比。肿瘤中基因cd274具有高mrna表达的患者(组iii+iv)与肿瘤中基因cd274具有低mrna表达的患者组i+ii相比,具有更加有利的临床进程。图7d显示了肿瘤中具有cd274高mrna表达和低甲基化(组iv)与高甲基化和低mrna表达(组ii)以及高甲基化和高mrna表达和低甲基化和低mrna表达(组i+iii)的患者的总生存期的kaplan-meier分析对比。可以看出,通过mrna表达分析和dna甲基化分析具有良好预后的组的患者(第iv组:高mrna表达和低甲基化)显示比通过mrna表达分析和dna甲基化分析具有不良预后组的患者(组ii:高甲基化和低表达)具有更显著有利的进程。所有其他患者(组i+iii)显示出在组ii和iv的预后之间的预后(图7d)。
图7详细地描述本发明的方法实施方案以基因cd274为例将mrna表达分析和dna甲基化分析组合用于改进恶性黑色素瘤患者的预后的判定。当将根据本发明的该方法应用于其他基因时,同样可以改善预后的确定。与cd274(图8a和图7d)相比,这也可以用于基因pdcd1lg2(图8b),pdcd1(图8c),btla(图8d),lag3(图8e),tigit(图8f),cd40(图8g),ctla4(图8h)和tnfrsf9(图8i)。图8显示了通过组合分析分析不同免疫调节基因的mrna表达分析和dna甲基化,回顾性地分析了470名具有已知预后的恶性黑色素瘤患者。类似于图7,患者的分组对应于每个基因相应的阈值。比较了肿瘤中高dna甲基化和低mrna表达(组ii)的患者、肿瘤中低dna甲基化和高mrna表达的患者(第iv组),以及其他患者(组i+iii)的疾病病程。对于所有研究的基因,可以表明组ii具有不利的疾病过程,组iv具有有利的疾病过程,而其余患者(组i+iii)具有中等的死亡风险。因此,第ii组代表高风险群体,第iv组代表低风险群体。
实施例8:基于免疫调节基因pdcd1lg2,cd274,pdcd1,btla,lag3,tigit,cd40,ctla4和tnfrsf9的dna甲基化分析和mrna的表达分析的组合确定恶性黑色素瘤患者的预后
在实施例3和4中,基于本发明已经证实患者基于总共九个不同的免疫检查点的基因(pdcd1lg2,cd274,pdcd1,btla,lag3,tigit,cd40,ctla4和tnfrsf9)的dna甲基化分析确定恶性黑素瘤的预后。确定恶性疾病患者的预后可以进一步通过组合各种基因的dna甲基化分析和mrna表达分析来改善。如实施例1和6中所述进行mrna表达和dna甲基化的测定。如实施例3和4中所述,对每种基因进行定量甲基化值的计算。首先基于9种基因的mrna表达将患者划分为预后组。第i组是基于9种基因的mrna表达可以被划分为良好预后的组。第i组包括肿瘤中显示9个基因中至少8个的mrna表达升高超过阈值的患者。第ii组是基于9个基因的mrna表达可被划分为不利预后的组。第ii组包含那些肿瘤中所分析的9个基因中,不到8个显示mrna表达高于阈值的患者。随后,根据9个基因的dna甲基化对患者进行分类。第iii组是基于9个基因的dna甲基化可以被划分为良好预后的组。iii组包括肿瘤9个基因中最多4个的dna甲基化显示高于阈值的患者。第iv组是基于9个基因的dna甲基化可以被划分为不良预后的组。iv组包括肿瘤9个基因中至少5个的dna甲基化显示高于阈值的患者。最终,基于9个基因的dna甲基化和mrna表达对患者分类。第v组是包括无论是mrna表达还是dna甲基化都具有良好预后的患者的组。第v组包括同时属于第i组和第iii组的患者。第vi组包括mrna表达和dna甲基化的预后都属于不良组的患者。第vi组包括第ii组和第iv组的患者。第vii组是包括或者是mrna表达或者是dna甲基化具有良好预后的患者的组。第vii组包括同时属于第i组和第iv组或第ii组和第iii组的患者。图9b示出基于所分析的9个基因中最多4个的dna甲基化具有不良预后的患者组,也就是相应的基因的甲基化高于阈值,总体来说显示出非常有利的预后(组iii)。而9个基因中至少5个的甲基化显示具有不良预后的患者组(第iv组),总体来说显示出非常不利的预后。图9a示出基于所分析的9个基因中至少8个的mrna表达具有良好预后的患者组(组i)比9个基因中少于8个的mrna表达具有良好预后的患者组(组ii),总体来说显示出更加有利的预后。基于dna甲基化对患者进行分组的阈值,例如可以设定以下甲基化值:pdcd1lg2为12.84%,cd274为13.11%,pdcd1为20.54%,tigit为86.41%,tnfrsf9为57.80%,lag3为66.94%,btla为91.31%,ctla4为13.10%,cd40为17.16%。基于mrna表达分析对患者进行分组的阈值,例如可以设定以下mrna表达值:pdcd1lg2为5.61,cd274为18.23,pdcd1为7.29,tigit为12.38,tnfrsf9为0.935,lag3为9.65,btla为3.33,ctla4为13.12,cd40为30.14。
在实施例7(图8)已经表明,根据本发明的dna甲基化分析和mrna表达分析的组合与或者仅基于dna甲基化分析,或者仅基于mrna表达分析相比,甚至能够更有区分性地判定恶性疾病患者的预后。因此如这里所示,如果分析免疫调节基因的dna甲基化分析和mrna表达分析的组合,本发明的这种有优势的效果甚至会进一步提高。图9c显示了患者的肿瘤无论是基于dna甲基化分析还是基于mrna表达分析,在当前研究的9个基因中在每个组中具有良好预后的患者(图9b中,第iii组和图9a中,第i组),总体预后非常好(图9c,第v组)。而无论是基于dna甲基化分析还是基于mrna表达分析,在当前研究的9个基因中在每个组中具有不良预后的患者(图9c,第vi组),总体预后不好。所有其他患者(图9c,第vii组)显示出比第vi组更有利,但不如第v组有利的进程。
实施例9:以免疫调节基因cd40和lag3为例通过启动子甲基化和基因内甲基化确定患有恶性黑素瘤的患者的预后
人们普遍认为基因的dna甲基化与mrna表达呈负相关。这意味着该基因的高甲基化与该基因的低mrna表达相关。在根据本发明的该示例性实施方案中,已经可以显示在基因内区域中也可以存在显著的正相关。这通过基因lag3和cd40的实例证明。如实施例1和6中所述进行mrna表达和dna甲基化的测定。如实施例4中所述进行两个基因lag3和cd40的启动子的相对甲基化的计算。为了确定基因内区域,使用下文所述的珠对并如实施例1中所述彼此进行对比,以获得每个基因座处的相对甲基化值。对于lag3基因内的dna甲基化分析,使用珠对cg11429292(seqidno:43)和cg14292870(seqidno:44),其例如可以分析seqidno:161和seqidno:350序列内的dna甲基化。对于cd40基因内的dna甲基化分析,使用珠对cg06218285(seqidno:50),其例如可以分析seqidno:297序列内的dna甲基化。
图10显示了根据本发明确定470名患有恶性黑素瘤的患者的预后的结果。基于单独地或组合使用启动子的甲基化分析和基因内区域的甲基化,回顾性地将患者分类。图10a和图10c显示位于启动子区域的cpg二核苷酸的相对甲基化与相应基因的mrna表达负相关。可以表明,这些基因的基因内区域与启动子区域相反,基因内区域的相对甲基化与mrna的表达显著正相关(图10b以及图10d)。图10e和10h显示了实施例4中已描述的基因lag3和cd40的dna甲基化分析的结果。
那些在lag3和cd40的启动子中具有升高的dna甲基化的患者(组ii)具有较差的进程(图10e和10h)。该关系在基因内区域令人惊讶地逆转。图10f中的组iii包含肿瘤的lag3基因的基因内甲基化超过20.67%的那些患者。第iv组包括那些肿瘤的甲基化低于20.67%的患者。lag3基因的基因内甲基化降低的那些患者具有不利的病程。基于基因lag3的基因内和启动子的甲基化的组合进行患者进程的kaplan-meier存活周期分析显示预后的确定被改进了(图10g)。第v组包括同时属于第ii组和第iv组的患者。第vii组包括同时属于第i组和第iii组的患者。第vi组包括或者属于ii组和iii组或者属于i组和iv组的患者。通过lag3基因的基因内甲基化和启动子甲基化显示出不利预后的患者形成了具有特别不利进程的组(图10g,组v)。
基于基因cd40的基因内和启动子的甲基化以及这两甲基化分析的组合(图10h,10i,10j)进行患者进程的kaplan-meier存活周期分析显示出与基因lag3的相似的结果。组i(图10h)包括肿瘤显示甲基化低于17.16%的那些患者。第ii组包括那些肿瘤显示甲基化高于17.16%的患者(图10h)。那些cd40启动子内的甲基化增高的患者具有不利的病程。第iii组(图10i)包括肿瘤显示甲基化高于35%的那些患者。第iv组(图10i)包括那些肿瘤显示甲基化低于35%的患者。那些cd40基因的基因内甲基化降低的患者具有不利的病程。第v组(图10j)包括属于第ii组和第iv组的那些患者。第vii组(图10j)包括属于第i组和第iii组的那些患者。第vi组包括ii组和iii组或i组和iv组的患者。结果表明,基于cd40的基因内和启动子的dna甲基化分析都具有良好预后的组中的患者形成具有非常良好预后的组。因此根据cd40的基因内和启动子的dna甲基化的组合与仅基于单一分析相比,能够甚至更有区分性的改善风险评估。
实施例10:通过cd274,pdcd1和pdcd1lg2的dna甲基化分析和mrna表达分析确定急性髓性白血病患者的预后
根据本发明的方法还可以确定患有恶性血液病的患者的预后。如实施例1和6中所述对182名急性髓性白血病患者的恶性细胞进行mrna表达分析和dna甲基化分析。基于dna甲基化分析的结果计算基因cd274,pdcd1和pdcd1lg2的相对dna甲基化,如实施例3中所述进行。
图11显示了182例急性髓性白血病患者的总存活期的kaplan-meier分析。出人意料的是,患者表现出基因cd274,pdcd1和pdcd1lg2的甲基化低于阈值(i组,图11a,11d,11g)的预后不良,而在前述实施例中,患者的这些免疫调节基因的甲基化增高显示不太有利的预测。因此,令人惊讶的发现是,根据本发明的方法还可以确定患有恶性血液病的患者的预后。基因cd274,pdcd1和pdcd1lg2的mrna表达的分析(图11b,11e,11h)也使得能够确定患者的预后。同样令人惊讶的是,即使在mrna表达的分析中,与先前描述的实施例7和8的结果相比,表达降低的那些患者(组iv)具有更有利的预后。图11c,11f和11i表明mrna表达和dna甲基化的组合分析与mrna表达或dna甲基化的单一分析相比,能够甚至更有区分性的改善风险评估。
实施例11:基于tnfrsf9,tigit,btla,havcr2,cd80,ctla4,icos,c10orf54,hhla2,cd160,kir2dl4和kir3dl1的dna甲基化分析确定急性髓系白血病的预后
基于本发明的方法使得能够还使用其它免疫调节基因确定恶性血液病患者的预后。对于dna甲基化分析,使用实施例10中收集的数据。基于如实施例4中描述的所述珠对数据确定tnfrsf9,tigit,btla和ctla4的相对甲基化。为了确定基因havcr2,cd80,icos,c10orf54,hhla2,cd160,kir2dl4和kir3dl1的相对甲基化,使用如实施例中1所提及的那些珠对,并用于计算所述的相对甲基化。对于havcr2,使用珠对cg09574807(seqidno:46),并能确定序列区域seqidno:286的甲基化。cd80的甲基化的确定是基于珠对cg12978275(seqidno:126)和cg13458803(seqidno:127),并能确定序列区域seqidno:163的甲基化。icos的dna甲基化的确定是基于珠对cg18561976(seqidno:112)和cg15344028(seqidno:113),并允许确定序列区域seqidno:92,seqidno:93和seqidno:95的甲基化。hhla2的dna甲基化的确定是基于四对珠对cg00915092(seqidno:122),cg14703454(seqidno:123),cg11326415(seqidno:124)和cg22926869(seqidno:125),并能确定序列区域seqidno:36,seqidno:195和seqidno:196的甲基化。对于基因c10orf54的序列区域seqidno:190和seqidno:191的dna甲基化分析,使用珠对ch.10.1529706r(seqidno:35)。对于基因cd160的序列区域seqidno:361,seqidno:362和seqidno:366的dna甲基化分析,使用珠对cg12832565(seqidno:150),cg10798745(seqidno:151)和cg15892497(seqidno:152)。对于基因kir2dl4的序列区域seqidno:217和seqidno:218的dna甲基化分析,使用珠对cg08326410(seqidno:143)。kir3dl1的dna甲基化分析是基于珠对cg15588997(seqidno:137),cg08129658(seqidno:138),cg02469067(seqidno:139),cg19689800(seqidno:140),cg06494497(seqidno:141)和cg05720980(seqidno:142),它们也能进行序列区域seqidno:230,seqidno:231和seqidno:232的甲基化分析。
图12显示了182例急性髓性白血病患者的总生存期的kaplan-meier分析。按照免疫调节基因tnfrsf9(图12a),tigit(图12b),btla(图12c),havcr2(图12d),cd80(图12e),ctla4(图12f),icos(图12g),c10orf54(图12h),hhla2(图12i),cd160(图12j),kir2dl4(图12k)和kir3dl1(图12l)的dna甲基化分析,对患者进行分组。二分法基于优化的阈值。阈值的优化按照这样的方式进行,将具有良好预后和不良预后的患者最佳的分离,也就是数秩检验(log-ranktest)中具有尽可能低的p值。所有分析的基因可以表明,恶性疾病的甲基化低于阈值的患者(组i)与甲基化高于阈值的患者相比,显示出不利的进程结果。
实施例12:通过cd274,pdcd1和pdcd1lg2的dna甲基化分析和mrna表达分析确定低级别胶质瘤患者的预后
根据本发明的方法还可以确定神经胶质瘤患者的预后。例如,在510名低级别神经胶质瘤患者中进行免疫调节基因cd274,pdcd1和pdcd1lg2的dna甲基化分析和mrna表达分析。如实施例1和6中所述进行dna和rna的制备,mrna表达分析和dna甲基化分析及其评估。如实施例3中所述,基于通过illuminahumanmethylation450beadchip产生的数据计算基因cd274,pdcd1和pdcd1lg2的相对甲基化。
实施例11中已经显示了血液恶性肿瘤,神经胶质瘤患者也可以基于dna甲基化分析、mrna表达分析或两个分析的组合来确定预后。图13显示,胶质瘤中免疫调节基因的甲基化低于阈值的患者(第i组)比甲基化高于阈值的患者(第ii组),具有显著更差的预后。这是可以由三种基因cd274(图13a),pdcd1(13d)和pdcd1lg2(图13g)同时显示。通过分析相应免疫调节基因的mrna表达,基于升高的基因表达也同样鉴定出该患者具有不良预后(组iii)。本发明尤其有利的是,根据mrna的表达分析和dna甲基化分析的组合,由此形成三个预后组,其分别是可能死亡的高风险(组i+iii),中等风险(组i+iv/ii+iii)和低风险(ii+iv组)。正如实施例10和11已经被证明的恶性血液病一样,在神经胶质瘤中有一个令人惊讶的发现,即与先前恶性疾病的分析相反,此处降低的mrna表达以及增加的dna甲基化与良好的预后相关。这强调,本发明能够确定患有各种恶性肿瘤的患者的预后。
实施例13:基于cd80,ctla4,icos和cd276的dna甲基化分析和mrna表达分析确定低级别胶质瘤患者的预后
根据本发明也能基于免疫调节基因cd80,ctla4,icos和cd276的dna甲基化分析和mrna表达分析确定低级别胶质瘤患者的预后。本分析基于如实施例12中所收集的甲基化和表达数据。如实施例11和4中所述进行基因cd80,ctla4和icos的相对dna甲基化的测定。为了确定cd276基因的相对dna甲基化,使用珠对cg24688248(seqidno:119),cg14910296(seqidno:120)和cg12524179(seqidno:121),并如实施例1中所述进行计算。这些珠对允许确定序列区域seqidno:181,seqidno:180和seqidno:183的dna甲基化。
图14显示了分析的510名低级别胶质瘤患者的总生存期的kaplan-meier分析。对患者基于免疫调节基因cd80(图14a,b,c),ctla4(图14d,e,f),icos(图14g,14h,14i),cd276(14j,14k,14l)的dna甲基化分析(图14a,14d,14g,14j),mrna的表达分析(图14b,14e,14h,14k),的dna甲基化和mrna表达分析的组合(图14c,14f,14i,14l)进行分组。对于基于dna甲基化的二分法,选择以下阈值:90.76%(cd80),86.43%(ctla4),84.75%(icos)和30.08%(cd276)。对于mrna表达的二分法,将中值用作阈值。组i包括具有甲基化低于阈值的患者,组ii由具有甲基化高于阈值的患者组成。iii组包括肿瘤的mrna表达高于中值的患者,而iv组的患者显示肿瘤中的mrna表达低于中值。如实施例12和图13中已经显示的cd274,pdcd1和pdcd1lg2,也可以基于基因cd80,ctla4,icos和cd276的dna甲基化和mrna表达以及两种分析的组合确定患者的预后。在这种情况下,令人惊讶的发现是,根据本发明,免疫调节基因的高dna甲基化和低mrna表达再次导致更有利的疾病进程。
实施例14:基于tnfrsf25,tnfrsf9,cd40,tigit,btla,havcr2,c10orf54,hhla2,lag3,cd160,kir2dl4和kir3dl1的dna甲基化分析确定低级别胶质瘤患者的预后
另外,也可以基于其他免疫调节基因的dna甲基化分析确定低级别胶质瘤患者的预后。在图15中所示的结果基于实施例12中描述的dna甲基化数据进行分析。用于确定基因tnfrsf9,cd40,tigit,btla,havcr2,c10orf54,hhla2,lag3,cd160,kir2dl4和kir3dl1的相对dna甲基化所使用的珠对以及dna甲基化的计算在实施例1,4和11中已经描述。例如,在该实施例中,为了确定基因tnfrsf25的dna甲基化,使用珠对cg27224823(seqidno:153),cg13331246(seqidno:154),cg23588699(seqidno:155)和cg10982045(seqidno:156),其可以确定序列区域seqidno:317,seqidno:318和seqidno:319的dna甲基化分析。也可以使用珠对cg00087884(seqidno:157),cg10059687(seqidno:158)和cg11756870(seqidno:159)为序列区域seqidno:317,seqidno:320和seqidno:32确定dna甲基化分析。
图15显示了510例低级别胶质瘤患者的总体存活期的kaplan-meier分析,其基于基因tnfrsf25(a),tnfrsf9(b),cd40(c),tigit(d),btla(e),havcr2(f),c10orf54(g),hhla2(h),lag3(i),cd160(j),kir2dl4(k)和kir3dl1(l)的dna甲基化分析进行回顾性分组。患者分组是基于基因的相对甲基化分成两组,使用以下阈值:61.88%(tnfrsf25),79.24%(tnfrsf9),43.24%(cd40),80.66%(tigit),84.64%(btla),2.495%(havcr2),66.52%(c10orf54),77.14%(hhla2),74.34%(lag3),92.89%(cd160),90.85%(kir2dl4)和52.28%(kir3dl1)。第i组包括dna甲基化低于阈值的患者;第ii组表示肿瘤显示dna甲基化高于阈值的患者。对于所有免疫调节基因,可以证明低dna甲基化与患者的不良预后相关。
实施例15:基于cd274,tnfrsf9,tigit,cd80,ctla4,cd276和hhla2的dna甲基化分析确定透明细胞肾细胞癌患者的预后
前述实施例已经表明,根据本发明的方法允许确定患者的各种恶性肿瘤,如腺癌,鳞状细胞癌,黑色素瘤,白血病和神经胶质瘤的预后。在该实施例中,将根据本发明的方法应用于患有透明细胞肾细胞癌的患者。如实施例1中所述产生dna甲基化数据。相对dna甲基化的计算使用基因cd274,tnfrsf9,tigit,cd80,ctla4,cd276和hhla2的珠对的数据,并按实施例3,4,11和13所述进行。
图16显示了318例透明细胞肾细胞癌患者的总体存活期的kaplan-meier分析,其基于免疫调节基因tigit(a),tnfrsf9(b),cd274(c),cd80(d),ctla4(e),cd276(f)和hhla2(g)的dna甲基化进行回顾性分组。每个基因的相对dna甲基化的二分值使用各自基因的所有患者的dna甲基化的中位数。组i包括肿瘤具有低于中值的dna甲基化的患者,而第ii组是肿瘤具有高于中值的dna甲基化的那些患者。图16示出了分析的7个基因中的6个具有增加的dna甲基化(组i)与不太有利的预后显著相关。令人惊讶的是,对于基因hhla2,如果免疫调节基因具有低于阈值的甲基化,则患有透明细胞肾细胞癌的患者的预后是不利的(图16g)。结果表明,对于除了那些在前面的实施例中描述的腺癌和扁状上皮细胞癌之外,本发明方法也适用于其它癌症。
实施例16:基于tnfrsf9,tigit,cd80,ctla4,cd276和hhla2dna甲基化分析与mrna的表达分析的组合确定透明细胞肾细胞癌患者的预后
本发明方法对各种恶性肿瘤的普遍适用性可以进一步通过318例透明细胞肾细胞癌患者的基因tnfrsf9,tigit,cd80,ctla4,cd276和hhla2的dna甲基化和mrna表达分析的组合来验证。dna甲基化分析和mrna表达分析如实施例1和6中描述的进行并评价。基因的相对甲基化的计算所使用的珠对如实施例4,11和13中描述。
图17示出了318例透明细胞肾细胞癌患者的总生存期的kaplan-meier分析,其基于基因tigit(a),tnfrsf9(b),cd80(c),ctla4(d),cd276(e)和hhla2(f)的dna甲基化和mrna表达的组合分析进行分组。二分法基于所有患者的甲基化或mrna表达的中值。第i组:甲基化低于阈值,mrna高于阈值;第iii组:甲基化高于中值,mrna低于中值;第ii组:甲基化高于阈值,mrna高于阈值或者甲基化低于阈值,mrna低于阈值。对于本实施例中分析的六个基因,显示患者可分为分别具有高,中,低死亡风险的三个组。基因tigit(a),tnfrsf9(b),cd80(c),ctla4(d)和cd276(e)的高dna甲基化和低mrna的表达(组i),与非常差的预后相关联,而对于基因hhla2(f)恰好呈现相反的关系,该基因的单独dna甲基化分析的令人惊讶的结果也已经在实施例15中证实了。
如在实施例13,12,10,8和7中已经证实的,根据本发明的免疫调节基因的dna甲基化和mrna表达的组合分析代表特别有利的方法实施方案,其比各单一分析能够获得区分效果更好的预后。
实施例17:通过pdcd1和cd274的dna甲基化分析确定恶性黑素瘤患者对免疫疗法的反应
本发明免疫调节基因的dna甲基化分析的用途为预测对免疫治疗的预测,其通过23名恶性黑色素瘤患者的组来验证。患者接受了帕母单抗的免疫治疗。该药物是针对由免疫调节基因pdcd1编码的pdcd1受体的单克隆抗体。该抗体通过与受体pdcd1相互作用,阻止cd274和pdcd1lg2编码的相应配体的结合,因此以这种方式,改变pdcd1以及cd274和/或pdcd1lg2的免疫调节作用。23例患者中的11例疾病逐渐恶化(患者13至23),11名患者(患者2至12)的疾病仍保持在稳定的水平或患者表现出肿瘤质量的减少。在1名患者(患者1)中,肿瘤回到目前为止已无法检测到的水平。
用于免疫调节基因pdcd1和cd274的dna甲基化分析的dna从肿瘤病人的皮肤转移,淋巴结转移和远处转移的细胞中提取。在开始免疫疗法之前手术采集转移灶。如实施例2中所述进行亚硫酸氢盐转化以用于转换dna。本发明的适于cd274和pdcd1免疫调节基因中每一个的dna甲基化分析采用如在实施例2和5中所述的定量实时pcr方法。
图18显示免疫调节基因cd274的患者依赖性的dna甲基化,该基因编码配体cd274,其结合免疫调节基因pdcd1编码的pdcd1受体。对帕母单抗治疗无反应的13至23名患者的肿瘤平均dna甲基化率为18.5%,而部分或完全反应的患者组(患者1至12)平均在肿瘤中仅显示6.2%的dna甲基化。
结果显示,以cd274基因的dna甲基化分析和药物帕母单抗为例,可以借助于根据本发明的方法预测对免疫疗法的反应(反应)。特别地,结果显示,例如,可以基于dna甲基化来选择患者,使得仅cd274基因的dna甲基化例如低于10%的患者可以接受帕母单抗治疗;而在具有高水平dna甲基化的患者中,在本实施例中对该治疗反应的可能性较低。在具有部分或完全反应的患者1至12中,10名(83%)具有低于10%的cd274的dna甲基化,因此在该实施例中可以适当地鉴定为对治疗具有预期反应的患者。总体而言,8名患者的cd274甲基化超过10%。对于这8名患者中的6名(75%),预测他们不会对免疫疗法做出反应是正确的。
该实施例表明了根据本发明的免疫调节基因的dna甲基化通常适合于预测对免疫疗法的反应。因此,如果免疫调节基因编码作为免疫检查点的配体,并用于确定药物,则能够确定,其能够改变配体的免疫调节作用,其中其抑制配体反应的受体。因此还可以显示根据本发明的免疫调节基因的dna甲基化分析也适用于预测对药物的反应,该药物拮抗地与另一种免疫调节基因编码的免疫检查点相互作用。
图19显示了根据本发明的免疫调节基因pdcd1的dna甲基化的相应测定。在显示出反应的患者1至12组中,在肿瘤样品中发现平均59%的pdcd1基因甲基化。这个甲基化值具有t检验为0.008的p值,并显著高于显示疾病恶化的患者的肿瘤的甲基化值,其平均甲基化值为22%。因此,pdcd1基因的dna甲基化水平也与对免疫治疗的反应相关,并且基于pdcd1的dna甲基化能够确定那些在开始治疗前可能对治疗有反应的患者。例如,12个具有反应的患者(患者1-12)中的9个(75%)显示基因pdcd1的dna甲基化超过25%,因此可以正确地识别为有反应的患者。通过免疫治疗肿瘤完全消退的病人(患者1)在所有样品的检查中目前具有最高的甲基化值98%,因此可以特别准确地识别。与此相反,11个不具有反应的患者(患者13-23)中的9个(82%)显示基因pdcd1的dna甲基化值低于25%,因此可以正确地识别为对免疫疗法帕母单抗没有反应的患者。这样的患者可以例如在将来可以不使用该帕母单抗免疫疗法,以避免其副作用。同时,通过更早地向这些患者应用另一种更有可能有效的疗法,可以获得宝贵的时间。例如,如果可以使用本发明的方法个性化选择另一种疗法以适应于编码其他免疫检查点的免疫调节基因的dna甲基化值,可以预估对药物的反应,该药物能够改变其他免疫检查点的免疫调节作用。
作为实例,图19还显示,考虑免疫调节基因的定量甲基化可以允许预测对治疗的反应。这允许患者在不应用阈值的情况下预测对治疗的反应。在本实施例中,肿瘤完全消退的患者1显示pdcd1基因的甲基化高于仅对治疗部分反应的患者2至12。
还可以组合应用根据本发明的各种免疫调节基因的dna甲基化分析,以便能够更精确地预测对免疫疗法的反应。从图18和19中可以看出,患者1显示肿瘤完全消退,其肿瘤中cd274的dna甲基化降低(3.6%),以及pdcd1的甲基化升高(98%)。cd274编码与pdcd1编码的受体结合的配体。帕母单抗是通过中断或抑制这种配体-受体结合来改变由这些基因编码的两个免疫检查点的免疫调节作用的药物。这一发现表明,本发明的几个免疫检查点的分析,特别是涉及相同的配体-受体的结合的那些,可以进一步提高对对免疫治疗的反应的预测。在本实施例中,这也可以通过至少部分地对治疗作出反应的12名患者(患者1至12)的事实来证明,所有的患者(100%)具有cd274甲基化低于10%或pdcd1的甲基化高于25%的特性。因此,可以正确地预测这些患者对免疫疗法的反应。11个不具有反应的患者(患者13-23)中的5个(45%)显示基因cd274的dna甲基化值高于10%并且基因pdcd1的dna甲基化值低于25%(患者14至16,19和21)。对于这些患者,通过pdcd1和cd274的组合dna甲基化分析可以特别可靠地预测对治疗没有反应。
实施例18使用不同免疫调节基因的dna甲基化分析和mrna表达分析确定头颈部鳞状细胞癌患者的预后
如实施例1中所述进行dna甲基化分析。对于表1中所描述的各个免疫调节基因的相对甲基化的计算使用表1中列出的infiniumhumanmethylation450beadchip芯片(illumina,inc.,sandiego,ca,usa)的珠对。基因组中这些珠对的靶序列同样在表1中由相应的seqidno(序列号)表示。对于珠对,计算在每种情况下每一个患者的相对甲基化,与实施例1所述类似,将珠对中特定磁珠的非甲基化和甲基化的水平进行对比。如实施例5,研究了528名头颈部鳞状细胞癌患者的总体存活期。
如实施例6中所述测定mrna表达。研究了表2中列出的mrna。
表1中总结的结果显示,根据本发明的免疫调节基因的dna甲基化与患者的存活期显著相关,因此可以确定患者的预后。风险比1由cox比例风险模型来确定和描述,其中描述了患者死亡风险的大小如何随着相应免疫调节基因的dna甲基化值的增加而增加。风险比大于1意味着患者的死亡风险随着dna甲基化水平的升高而增加。风险比小于1会降低这种风险。
本实施例显示,根据本发明表1中列出的免疫调节基因的dna甲基化分析允许患者预后的确定。此外,该实施例表明预后的确定不仅在于能够基于甲基化的阈值将患者划分为不同的预后组,而且在于根据统计模型,例如cox比例风险模型,可以确定患者预后的程度依赖于甲基化值的变化程度。
在表1中基于基因实例adora2a,btnl2,c10orf54,cd160,cd276,cd48,cd80,cd86,pdcd1,tigit和tnfrsf18也表明,一个基因内的不同区域对于本发明方法的使用也是合适的。这由以下事实证实:在这些基因中,通过将多个区域用不同的珠对检测进行dna甲基化分析,每个都能成功确定预后。
表1中的风险比2描述了,肿瘤中甲基化值高于阈值的患者与值低于阈值的患者相比,其死亡风险的程度更高。令人惊讶的是,例如,在免疫调节基因cd274为例的情况下,两个区域seqidno:425和seqidno:426都显示增加的甲基化与降低的死亡风险相关联,而在实施例3中cd274基因的其它区域表明,增加的甲基化与恶性黑色素瘤患者的不良存活率相关。cd276也具有类似的发现,其中seqidno:427,seqidno:428和seqidno:429与存活期呈负相关,也就是说这些区域的甲基化增加与死亡风险降低相关。相反,seqidno:430区域的甲基化与死亡风险正相关。因此,它表示本发明的dna甲基化分析的一个特别的优点在于可以通过选择基因中一个或几个研究区域来实现区分效果更好的预后,如本实施例,可以单独考虑mrna表达或蛋白表达。
表1:528名头颈部鳞状细胞癌患者的总体存活分析。研究了所提及的基因的dna甲基化与患者的存活期之间的关系。在表中多次列出的基因中,进行了根据本发明根据相应的珠对的甲基化测定,所述珠对覆盖相应基因不同区域。这些区域由具有相应seqidno的序列确定。风险比1指的是甲基化值没有事先进行二分,风险比2描述了甲基化高于阈值的患者相对于甲基化低于阈值的患者具有更高的死亡风险。p值1和2分别表示风险比1和2。
表2显示了上述实施例的免疫调节基因的mrna的表达分析也能够预测患者的生存期,因此,可以补充本发明的dna甲基化分析。根据示例性阈值对患者进行分组。该阈值是免疫检查点的mrna分子的数量,相对于所有由rna-seq分析的mrna分子(归一化计数),通过该值将患者分为具有良好和不良预后的组特别好。肿瘤中cd274或cd276的mrna表达高于相应阈值的患者与肿瘤中这些基因低于相应阈值的患者相比,具有更加显著不利的进程。而对于基因adora2a,btnl2,c10orf54,cd160,cd27,cd48,pdcd1,tigit,tnfrsf18,tnfrsf25和tnfrsf4,肿瘤中上述基因高于相应阈值的患者相反地具有更加有利的预后。因此,通过该实施例可以证明mrna表达分析可以可靠地用作根据本发明确定预后的另一指标。
表2:528名头颈部鳞状细胞癌患者的总体存活分析。研究了所述免疫调节基因的mrna表达与患者存活期之间的关系。通过基于阈值的二分mrna表达值确定风险比。
实施例19:基于免疫调节基因的共甲基化和共表达确定dna甲基化和mrna表达
如实施例8中已经示出的,免疫调节基因通常共表达,即,例如恶性疾病细胞和/或免疫细胞,如t淋巴细胞不仅单个免疫调节基因表达,而且几个免疫调节基因同时表达。在该实验中,多种免疫调节基因的mrna表达和dna甲基化的测定如实施例1和6所描述。生成了头颈部肿瘤患者样品中的520例mrna表达数据和528例dna甲基化数据。
表3示显示是,对于示例性示出的免疫调节基因,其与肿瘤中各种免疫调节基因的mrna表达具有统计学意义上的显著正相关。因此,可以基于单个或多个免疫调节基因的mrna表达的测定推导出其他免疫调节基因的mrna表达。
表3:520名头颈部癌症患者肿瘤中不同免疫调节基因的mrna表达的spearman等级相关性(spearman’sρ)。除icoslg与cd274(p=0.008)以及icoslg与pdcd1lg2(p=0.070)的相关性外,其余所有相关性均显示显著性水平p<0.001。
pdcd1lg2和cd274编码配体,其都与基因pdcd1编码的受体结合。检查了这两个基因彼此的dna甲基化的相关性。对于pdcd1lg2的dna甲基化分析,使用珠对cg07211259(seqidno:31)。对于cd274,使用珠对cg15837913(seqidno:8),cg13474877(seqidno:11)和cg19724470(seqidno:12)。相关性是由spearman等级相关性来确定,相关系数用spearman’sρ表示。统计显著性表示为p值。珠对cg07211259的结果与珠对cg15837913(ρ=0.33,p<0.001)相关,cg07211259与cg13474877(ρ=0.24,p<0.001)相关,cg07211259与cg19724470(ρ=0.42,p<0.001)相关。这表明,例如基于所建立的相关性,通过pdcd1lg2的dna甲基化分析可以对cd274的甲基化作出预测。以这种方式,例如可以基于pdcd1lg2的dna甲基化分析来预测对于一种药物疗法的反应,该药物抑制由cd274或pdcd1编码的免疫检查点,例如由于所示的相关性,pdcd1lg2的dna甲基化分析可以指示cd274的特定的dna甲基化,这很可能使得对所述药物疗法具有反应。
- 还没有人留言评论。精彩留言会获得点赞!