尿嘧啶化合物晶体的制造方法与流程
本申请对于2016年7月15日申请的日本专利申请第2016-140053号、2016年12月22日申请的日本专利申请第2016-248827号、以及2017年3月22日申请的日本专利申请第2017-055556号主张优先权及它们的利益,通过参照将它们的全部内容引入本申请中。本发明涉及一种含有作为除草剂的有效成分的尿嘧啶化合物的晶体的制造方法。
背景技术:
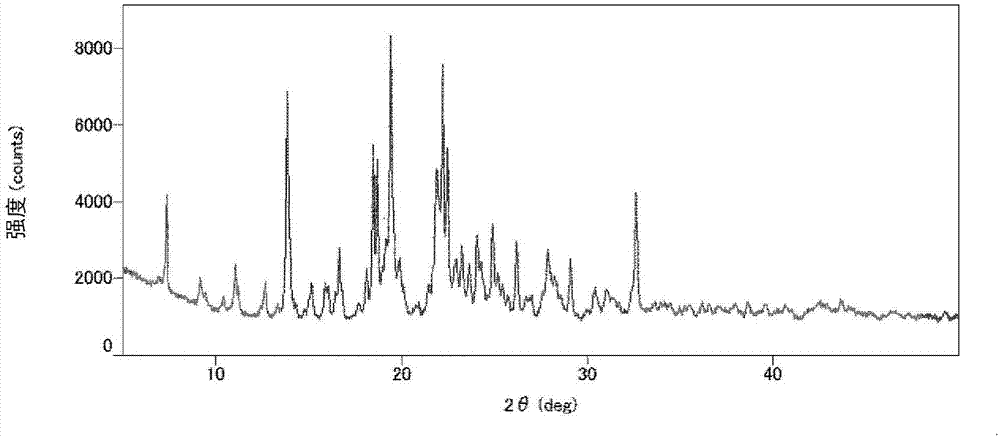
:作为除草剂的有效成分已知有以下述的式(1)表示的尿嘧啶化合物(以下记作本尿嘧啶化合物)(专利文献1;化合物7-8)。另外,记载过经由多阶段的反应工序制造本尿嘧啶化合物(专利文献1至5等)。现有技术文献专利文献专利文献1:日本特开2002-155061号公报专利文献2:日本特开2003-48885号公报专利文献3:日本特开2003-286284号公报专利文献4:日本特开2003-286285号公报专利文献5:日本特开2003-321468号公报技术实现要素:发明所要解决的问题在经过多阶段的反应工序以工业化规模制造农药活性成分等有机化合物的情况下,重要的是确立以高纯度取得最终化合物的提纯方法。本发明的目的在于,提供一种利用能够以工业化规模实施的操作获得纯度高的本尿嘧啶化合物晶体的制造方法。用于解决问题的方法本发明人等对以高纯度取得以式(1)表示的本尿嘧啶化合物的方法进行了深入研究,结果发现,在将含有本尿嘧啶化合物的组合物(该组合物可以以任意的纯度含有本尿嘧啶化合物;以下记作粗尿嘧啶组合物)溶解于包含特定的有机溶剂的溶剂中后,使晶体析出,由此能够以高纯度得到本尿嘧啶化合物晶体,从而完成了本发明。本发明的本尿嘧啶化合物晶体的制造方法即为:[1]一种本尿嘧啶化合物晶体的制造方法,其特征在于,将粗尿嘧啶组合物溶解于包含c3-c6醇溶剂及芳香族溶剂的有机溶剂中,使本尿嘧啶化合物的晶体从溶液中析出。[2]根据[1]记载的本尿嘧啶化合物晶体的制造方法,其中,使本尿嘧啶化合物的晶体析出的方法是通过冷却粗尿嘧啶组合物的溶液来进行的。[3]根据[1]或[2]记载的本尿嘧啶化合物晶体的制造方法,其中,c3-c6醇溶剂为选自1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、1-戊醇、3-甲基-1-丁醇及2-甲基-2-丁醇中的一种或多种的混合物,芳香族溶剂为选自甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯及氯苯中的一种或多种的混合物。[4]根据[1]、[2]或[3]中任一项记载的本尿嘧啶化合物晶体的制造方法,其中,c3-c6醇溶剂与芳香族溶剂的重量比为50:50~98:2。发明效果根据本发明,在经过多阶段的反应工序制造的本尿嘧啶化合物中,能够以高纯度取得作为最终化合物的本尿嘧啶化合物晶体。附图说明图1是表示利用本发明的制造方法得到的本尿嘧啶化合物晶体的粉末x射线衍射的一例的图。具体实施方式本发明中使用的包含两种有机溶剂的混合物中,一方为c3-c6醇溶剂(以下记作a溶剂),另一方为芳香族溶剂(以下记作b溶剂)。a溶剂为以式roh(r为c3~c6的烃基)表示的溶剂,具体而言可以举出1-丙醇、2-丙醇(异丙醇)、1-丁醇、2-丁醇(仲丁醇)、2-甲基-1-丙醇(异丁醇)、2-甲基-2-丙醇(叔丁醇)、1-戊醇、3-甲基-1-丁醇(异戊醇)、2-甲基-2-丁醇(叔戊醇)、1-己醇及环己醇等醇溶剂。b溶剂是作为可以由一个或多个卤素原子(例如氯原子)取代的苯的溶剂、或者是作为可以由一个或多个c1~c3的脂肪族烃基取代的苯的溶剂,所述一个或多个c1~c3的脂肪族烃基可以由该一个或多个卤素原子取代,优选为作为可以由一个或多个c1~c3的脂肪族烃基或一个或多个氯原子取代的苯的溶剂,具体而言可以举出苯、甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯、丙苯、异丙苯、1,3,5-三甲基苯(均三甲苯)、1,2,4-三甲基苯(假枯烯(pseudocumene))、氯苯、邻二氯苯、间二氯苯及对二氯苯等芳香族溶剂。本发明的方法中所用的本尿嘧啶化合物例如为利用以下记载的式(2)至式(5)制造的化合物。式(2)至式(5)中,以式(a1)表示的化合物(以下记作化合物(a1))及以式(a11)表示的化合物(以下记作化合物(a11))是日本特开2002-363170、国际公开wo2007/083090等中记载的化合物,可以利用该文献中记载的方法来制造。粗尿嘧啶组合物含有60~97重量%的本尿嘧啶化合物,并且含有式(2)至式(5)的制造中间体、来源于该制造中间体等的衍生物等化合物(以下记作本混入化合物。)或焦油成分。作为本混入化合物的具体例,可以设想为式(2)至式(5)的化合物(a1)、以式(a3)表示的化合物(以下记作化合物(a3))、以式(a4)表示的化合物(以下记作化合物(a4))、以式(a5)表示的化合物(以下记作化合物(a5))、以式(a8)表示的化合物(以下记作化合物(a8))、以式(a9)表示的化合物(以下记作化合物(a9))、以式(a10)表示的化合物(以下记作化合物(a10))、化合物(a11)等室温下为固体的制造中间体、来源于该制造中间体的衍生物等。本发明中,粗尿嘧啶组合物与有机溶剂(a溶剂及b溶剂)的比率以粗尿嘧啶组合物中的本尿嘧啶化合物的纯成分为基准。本发明的制造方法中,本尿嘧啶化合物晶体的析出时的溶剂(合计)的量相对于本尿嘧啶化合物的纯成分1重量份为1.5~20重量份,例如为2~20重量份。本发明中,作为从粗尿嘧啶组合物的溶液中析出本尿嘧啶化合物晶体的方法,可以使用通过将该溶液冷却而进行的冷却晶析、或通过慢慢地添加本尿嘧啶化合物的溶解度低的溶剂而进行的不良溶剂添加晶析。在任意一种方法中,本尿嘧啶化合物晶体析出的温度都为-10~80℃,优选为-10~50℃的范围。a溶剂及b溶剂可以是从各自的组中选择的一种溶剂,也可以是从各自的组中选择的多种溶剂的混合。在将粗尿嘧啶组合物溶解于有机溶剂中时,可以向a溶剂与b溶剂的混合物中添加粗尿嘧啶组合物,也可以在使粗尿嘧啶组合物溶解于a溶剂中后添加b溶剂。a溶剂与b溶剂的比率在本尿嘧啶化合物析出的时刻以重量比计为a溶剂:b溶剂=10:90~99:1,优选为50:50~98:2,更优选为60:40~98:2。对从溶剂中析出的本尿嘧啶化合物利用常法进行过滤、清洗,以湿滤饼的形式从母液中分离。在将该湿滤饼干燥的情况下,可以采用使用了氮气或氦气等非活性气体的通气干燥、加热干燥、减压干燥、或将它们组合了的干燥等一般的方法来进行。本发明中,作为优选的方式可以举出以下的方式。作为a溶剂,优选选自1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、1-戊醇、3-甲基-1-丁醇及2-甲基-2-丁醇中的一种或多种混合物。尤其优选2-丙醇。作为b溶剂,优选选自甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯及氯苯中的一种或多种混合物。尤其优选选自邻二甲苯、间二甲苯、对二甲苯、以及乙苯中的一种或多种混合物。在a溶剂为2-丙醇的情况下,a溶剂与b溶剂的比率以重量比计优选为2-丙醇:b溶剂=80:20~98:2的范围,更优选为85:15~98:2的范围。a溶剂与b溶剂的合计量的优选范围根据粗尿嘧啶组合物中的本尿嘧啶化合物的含量而不同,然而通常相对于粗尿嘧啶组合物中的本尿嘧啶化合物的纯成分1重量份,优选为2~8重量份,更优选为3~8重量份。本发明中,粗尿嘧啶组合物的制造方法没有限定,具体而言可以举出利用上述的式(2)中记载的方法制造的粗尿嘧啶组合物。在本发明中,还包括以下的本尿嘧啶化合物晶体的制造方法。一种本尿嘧啶化合物晶体的制造方法,该制造方法包括:使化合物(a1)与三氟乙酰乙酸烷基酯反应的工序(工序1);使包含工序1中得到的化合物(a3)的粗产物与氰酸盐在质子酸的存在下反应的工序(工序2);使包含工序2中得到的化合物(a4)的粗产物与甲基化剂在碱的存在下反应的工序(工序3);以及将包含工序3中得到的本尿嘧啶化合物的粗产物溶解于包含c3-c6醇溶剂及芳香族溶剂的有机溶剂中、并使本尿嘧啶化合物的晶体从溶液中析出的工序(工序4)。作为工序1中的三氟乙酰乙酸烷基酯,可以举出4,4,4-三氟乙酰乙酸乙酯及4,4,4-三氟乙酰乙酸甲酯。作为工序2中的氰酸盐,可以举出氰酸钾及氰酸钠。作为质子酸,可以举出有机酸(例如乙酸、丙酸、丁酸、苯甲酸、对甲苯磺酸、以及甲磺酸)、以及无机酸(例如盐酸及硫酸)。作为工序3中的甲基化剂,可以举出碘甲烷、溴甲烷及硫酸二甲酯。作为碱,可以举出三乙基胺、二异丙基乙基胺、碳酸钾及碳酸钠。作为图1给出利用本发明的制造方法得到的本尿嘧啶化合物晶体的粉末x射线衍射的测定结果的一例。在一个实施方式中,本尿嘧啶化合物晶体在利用cu-kα射线的粉末x射线衍射中,具有以表1表示的衍射峰。[表1]即,在一个实施方式中,本尿嘧啶化合物晶体是在利用cu-kα射线的粉末x射线衍射中主要在2θ=7.4±0.2°、13.8±0.2°、17.7±0.2°、18.5±0.2°、18.7±0.2°、19.4±0.2°、21.8±0.2°、22.2±0.2°、22.5±0.2°、24.0±0.2°、24.9±0.2°、26.2±0.2°及32.6±0.2°具有衍射峰的晶体。粉末x射线衍射的条件如下所示。(测定条件)粉末x射线衍射装置:smartlab(株式会社rigaku制)x射线输出功率:cukα、45kv、200ma取样宽度:0.02°扫描范围:5°~50°[实施例]以下,利用实施例对本发明进行详细说明。实施例中,没有特别记载时的含量是利用使用了高效液相色谱的绝对标准曲线法测定。参考制造例1在式(2)的制造路线中,制备出粗尿嘧啶组合物。需要说明的是,在第一工序及第二工序中均未进行硅胶柱色谱等提纯操作,使用各工序中得到的粗产物作为下一工序的原料。实施例1向烧瓶内,加入参考制造例1中得到的含有本尿嘧啶化合物的组合物126.6重量份(本尿嘧啶化合物的含量:79.0%。作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇(关东化学株式会社制)316.5重量份、以及二甲苯(住友商事chemical株式会社制;邻二甲苯、间二甲苯、对二甲苯及乙苯的混合物)64.6重量份,在氮气气氛下加热至70℃。确认没有不溶物后,一边搅拌该混合物一边慢慢地冷却,确认在35℃开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤分离。将该晶体用冰浴了的2-丙醇(同上)与二甲苯(同上)的5:1(重量比)混合溶剂253.2重量份清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体74.7重量份。所得的本尿嘧啶化合物晶体中的本尿嘧啶化合物的含量为96.4%。以下,给出析晶前的组合物及所得的晶体的利用高效液相色谱的分析结果。可以确认所得的晶体的本尿嘧啶化合物的纯度高,化合物(a1)、化合物(a3)的含量得到减少。[表2]面积百分率析晶前的组合物所得的晶体本尿嘧啶化合物85.6%97.6%化合物(a3)0.9%0.1%化合物(a1)1.1%0.2%※液相色谱的测定条件装置岛津lc-20a、hplc、色谱柱sumipaxodsz-clue(3μm4.6mmφ×100mm)、流动相a液:0.1%磷酸水溶液、b液:乙腈、b液的比例:10%(0min)-[30min]→90%(5min)→10%(15min)、流量1ml/min、温度40℃、检测波长274nm将如下所述的本尿嘧啶化合物或制造中间体的类似化合物添加到粗尿嘧啶组合物中,用于本发明的制造方法,由此可以确认本发明的制造方法对于化合物(a1)、化合物(a3)以外的化合物也有优异的提纯效果。实施例2将分别包含0.1~1%的下述的参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇300重量份、以及二甲苯65重量份的混合物加热到70℃。将该混合物慢慢地冷却,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用2-丙醇与二甲苯的5:1(重量比)混合溶剂清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。将向实施例2中记载的粗尿嘧啶组合物中添加的化合物的制造方法记载如下。参考制造例2将化合物(a11)(1.0mmol)与4,4,4-三氟乙酰乙酸乙酯(1.0mmol)溶解于甲苯10ml中,将该混合物在加热回流条件下搅拌3小时。将回流液通过填充有分子筛5a的柱子而使之吸附乙醇。将反应溶液冷却后,得到以式(a12)表示的化合物(以下记作化合物(a12))。向化合物(a12)(0.8mmol)与乙酸3ml的混合物中,加入氰酸钾(0.8mmol),在50℃搅拌1小时,再在110℃搅拌2小时。将反应混合物冷却后,注入水中,用乙酸乙酯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(a13)表示的化合物(以下记作化合物(a13))。向化合物(a13)(0.5mmol)与碳酸钾(0.7mmol)与乙腈5ml的混合物中,在室温下加入硫酸二甲酯(0.7mmol),在50℃搅拌1小时。将反应混合物冷却后,注入水中,用甲苯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(b1)表示的化合物(以下记作化合物(b1))。将化合物(b1)的物性值表示如下。1h-nmr(cd3cn-d2o)δ:7.93(1h,dd,j=4.9,1.5hz),7.45(1h,dd,j=7.6,1.5hz),7.26-7.24(1h,m),7.07-7.06(1h,m),7.03-7.02(1h,m),6.93-6.92(1h,m),6.33(1h,s),4.85-4.84(2h,m),4.14(2h,q,j=7.1hz),3.43(3h,s),1.19(3h,t,j=7.1hz).参考制造例3将以式(a21)表示的化合物(以下记作化合物(a21))(1.0mmol)和4,4,4-三氟乙酰乙酸乙酯(1.0mmol)溶解于甲苯10ml中,将该混合物在加热回流条件下搅拌3小时。将回流液通过填充有分子筛5a的柱子而使之吸附乙醇。将反应溶液冷却后,得到以式(a22)表示的化合物(以下记作化合物(a22))。向化合物(a22)(0.8mmol)与乙酸3ml的混合物中,加入氰酸钾(0.8mmol),在50℃搅拌1小时,继而在110℃搅拌2小时。将反应混合物冷却后,注入水中,用乙酸乙酯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(a23)表示的化合物(以下记作化合物(a23))。向化合物(a23)(0.5mmol)与碳酸钾(0.7mmol)与乙腈5ml的混合物中,在室温下加入硫酸二甲酯(0.7mmol),在50℃搅拌1小时。将反应混合物冷却后,注入水中,用甲苯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(b2)表示的化合物(以下记作化合物(b2))。参考制造例4将以式(a31)表示的化合物(以下记作化合物(a31))(1.0mmol)和4,4,4-三氟乙酰乙酸乙酯(1.0mmol)溶解于甲苯10ml中,将该混合物在加热回流条件下搅拌3小时。将回流液通过填充有分子筛5a的柱子而使之吸附乙醇。将反应溶液冷却后,得到以式(a32)表示的化合物(以下记作化合物(a32))。向化合物(a32)(0.8mmol)与乙酸3ml的混合物中,加入氰酸钾(0.8mmol),在50℃搅拌1小时,继而在110℃搅拌2小时。将反应混合物冷却后,注入水中,用乙酸乙酯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(a33)表示的化合物(以下记作化合物(a33))。向化合物(a33)(0.5mmol)与碳酸钾(0.7mmol)与乙腈5ml的混合物中,在室温下加入硫酸二甲酯(0.7mmol),在50℃搅拌1小时。将反应混合物冷却后,注入水中,用甲苯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(b3)表示的化合物(以下记作化合物(b3))。参考制造例5将以式(a41)表示的化合物(以下记作化合物(a41))(1.0mmol)与4,4,4-三氟乙酰乙酸乙酯(1.0mmol)溶解于甲苯10ml中,将该混合物在加热回流条件下搅拌3小时。将回流液通过填充有分子筛5a的柱子而使之吸附乙醇。将反应溶液冷却后,得到以式(a42)表示的化合物(以下记作化合物(a42))。向化合物(a42)(0.8mmol)与乙酸3ml的混合物中,加入氰酸钾(0.8mmol),在50℃搅拌1小时,继而在110℃搅拌2小时。将反应混合物冷却后,注入水中,用乙酸乙酯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(a43)表示的化合物(以下记作化合物(a43))。向化合物(a43)(0.5mmol)与碳酸钾(0.7mmol)与乙腈5ml的混合物中,在室温下加入硫酸二甲酯(0.7mmol),在50℃搅拌1小时。将反应混合物冷却后,注入水中,用甲苯萃取。将有机层用饱和食盐水清洗、浓缩,对残渣实施柱色谱处理,得到以式(b4)表示的化合物(以下记作化合物(b4))。参考制造例6将化合物(a4)(1.0mmol)溶解于甲苯5ml中,在室温下滴加重氮甲烷(1.0mmol)的甲苯溶液1ml。其后,将反应液注入水中,用甲苯萃取。将有机层用饱和食盐水清洗、浓缩。对残渣实施硅胶色谱处理,得到以式(b9)表示的化合物(以下记作化合物(b9))。lc/ms(esi-ms(posi)):518[m+h]+参考制造例7向化合物(a4)0.71g、甲苯50ml、以及甲醇10ml的混合物中,在冰浴下滴加三甲基甲硅烷基重氮甲烷的己烷溶液(0.6mol/l)4.7ml。将该混合物在相同温度下搅拌30分钟后,加入乙酸,直至发泡消失。将反应混合物浓缩后,对所得的残渣实施硅胶色谱处理,得到化合物(b9)0.13g。1h-nmr(cdcl3)δ:7.91(1h,dd,j=5.0,1.6hz),7.39―7.34(2h,m),6.97―6.93(2h,m),6.53(1h,s),5.02(1h,d,j=16.0hz),4.84(1h,d,j=16.0hz),4.15(2h,q,j=7.1hz),3.94(3h,s),1.24(3h,t,j=7.1hz).参考制造例8将化合物(a1)(1.0mmol)和三乙基胺(1.5mmol)溶解于四氢呋喃8ml中,在室温下加入乙酰氯(1.5mmol)。将该混合物在40℃搅拌后,将反应溶液注入水中。用乙酸乙酯萃取,将有机层用饱和食盐水清洗、浓缩。对所得的残渣实施柱色谱处理,得到以式(b10)表示的化合物(以下记作化合物(b10))。lc/ms(esi-ms(posi)):383[m+h]+实施例3将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇169重量份、以及二甲苯4重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例4将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇160重量份、以及二甲苯18重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例5将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇255重量份、以及二甲苯28重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例6将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇141重量份、以及二甲苯36重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例7将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇256重量份、以及甲苯29重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例8将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、1-丁醇255重量份、以及二甲苯27重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的1-丁醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。实施例9将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、2-丙醇255重量份、以及氯苯29重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的2-丙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。对所得的晶体进行分析,可以确认本尿嘧啶化合物的纯度高,本尿嘧啶化合物以外的化合物的量得到减少。比较例1将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、乙醇271重量份、以及二甲苯11重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的乙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。比较例2将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、乙醇281重量份、以及二甲苯2重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的乙醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。比较例3将分别含有0.1~1%的上述参考制造例2~8中制备的化合物的粗尿嘧啶组合物(作为本尿嘧啶化合物的纯成分为100重量份)、甲醇270重量份、以及二甲苯10重量份的混合物加热到60℃。慢慢地冷却该混合物,在50℃以下的温度开始晶体析出。进一步冷却到0℃后,将产生的晶体利用过滤加以分离。用0℃的甲醇清洗。将清洗后的晶体在减压条件下干燥,得到本尿嘧啶化合物晶体。产业上的可利用性利用本发明的制造方法,可以取得高纯度的本尿嘧啶化合物晶体。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1