用于可逆地保护生物分子的试剂的制作方法
本发明涉及用于可逆保护生物分子的试剂。特别地,其涉及衍生自氮杂靛红酸酐的化合物及其用于保护生物分子、特别是酶以阻断其活性的用途。本发明还涉及以这种方式保护的生物分子和使用这些试剂的方法。
背景技术:
利用分子生物学技术对生物样品中存在的核酸靶标进行特异、快速和定量检测的现代体外诊断测试最常使用能够从特定引物扩增核酸靶标的聚合酶。
已经开发了所谓的“热启动”聚合酶,因为它们与其天然版本相比具有许多优点,特别是在诊断测试中提供了更好的灵敏度性能。
“热启动”聚合酶的操作原理是通过抗体、适体或化学试剂暂时阻断其活性,然后在pcr扩增期间一旦温度升高就恢复其活性。
由于其低成本,用化学试剂阻断是特别有利的。在现有技术中已经描述了通过酰胺化赖氨酸残基的胺官能团使聚合酶失活(例如参见:enzymeandmicrobialtechnology36(2005)947-952,ariellouwrier,annevandervalk的“thermallyreversibleinactivationoftaqpolymeraseinanorganicsolventforimplementationinhotstartpcr”)。在第一个pcr循环期间95℃的变性步骤中或在高温下的预活化步骤中,切割保护基团并恢复其酶活性。
专利us5677152和us5773258公开了柠康酐及其衍生物在水性介质中用于临时保护taq聚合酶的赖氨酸的nh2基团的用途。然后,在用于pcr反应的tris缓冲液存在下,通过热处理后酸化的介质来水解酸酐和胺反应后产生的酰胺。
还已经证明,靛红酸酐衍生物能够与亲核基团如蛋白质上存在的胺反应。1982年,moormana.r.等人描述了靛红酸酐对蛋白质、胰凝乳蛋白酶的酰化作用(moormana.r.,abelesr.h.j.,am.chem.soc.,1982,104,6785-6786)。该反应通过产生稳定的氨茴酸衍生物导致蛋白质失活。最近,hooker等人使用靛红酸酐的反应性来修饰溶菌酶分子的赖氨酸残基,从而引入苯胺结构,然后通过氧化偶联将其功能化(hookerj.m.,esser-kahna.b.,francism.b.,j.am.chem.soc.,2006,106,1558-1559)。由此,通过靛红酸酐酰化存在于赖氨酸侧链上的胺基导致形成非常稳定的酰胺键。
专利申请fr1257526描述了衍生自靛红酸酐的酰化剂,其用于核糖核酸(rna)或嵌合核酸(rna/dna)的官能化、标记、捕获或分离。出于将感兴趣的基团固定在这些生物分子上的目的,更具体地描述了酰化剂。
本发明提出提供用于可逆保护生物分子新试剂,特别是用于制备“热启动”酶的新试剂,所述“热启动”酶是指那些通过热处理可以容易去保护的酶,所述新试剂优选具有以下优点中的一种或多于一种:
-它们便宜,
-它们很稳定,
-它们可溶于水性介质,
-它们在反应速度和去保护速度方面均高效,
-它们与生物分子反应的产物稳定,
-它们不需要使用tris缓冲液进行热去保护,
和/或
-它们不会产生不良副反应。
因此,本申请中描述的试剂特别适合于临时灭活参与核酸聚合的酶,例如taq聚合酶或某些逆转录酶,更通常为用于体外诊断技术的酶。
技术实现要素:
因此,本发明涉及下式(i)的化合物:
其中
x是共价键或c1-c4烷基,
y是亲核基团,优选o、s、nr4、o-nr4、nh-o或nh-nr4,r4是h或c1-c4烷基,
z1、z2、z3各自独立地表示n或c,优选地z3表示c,更优选地z3为c且r3位于z3,
r1为h、经取代或未经取代的c1-c6烷基、经取代或未经取代的链烯基、经取代或未经取代的芳基、或经取代或未经取代的杂环,优选地r1为甲基或乙基。
r2是热不稳定和/或酸不稳定的保护基团,
r3是h、经取代或未经取代的c1-c12烷基,例如异丙基、异丁基、仲丁基、叔丁基、异戊基或2,2-二甲基丙基,经取代或未经取代的芳基,经取代或未经取代的杂环,酰基,取代经或未经取代的链烯基,卤素(例如f、cl、br和i)或氰基。
在优选的实施方案中,根据本发明的化合物是下式(ii):
其中x、y、z1、z2、r1、r2和r3如上文对式(i)所定义的。
在一个具体实施方案中,上述式(i)和(ii)中的热不稳定和/或酸不稳定基团r2选自叔丁氧基羰基(boc)、经取代或未经取代的苯氧基乙酰基、三苯甲基、甲氧基三苯甲基、二甲氧基三苯甲基或柠康酰基。
在可以与前述实施方案组合的另一具体实施方案中,r1为甲基。
在可以与前述实施方案组合的另一个具体实施方案中,r3为碘。
在可以与前述实施方案组合的另一特定实施方案中,z1为n,z2为c。
特别地,本发明涉及具有以下结构的化合物之一:
本发明还涉及制备经保护的生物分子的方法,其包括将如上所述的本发明化合物与包含一个或多于一个亲核基团的生物分子接触以形成经保护的生物分子,其条件是所述生物分子的一个或多于一个亲核基团能够酰化。
在特别优选的实施方案中,生物分子包含胺官能团。特别地,生物分子是包含亲核官能团的蛋白质,特别是包含其赖氨酸残基或末端氨基酸的胺官能团、丝氨酸的醇官能团、和/或半胱氨酸的硫醇官能团的蛋白质。
本发明特别涉及由下式(iii)表示的经保护的生物分子:
其中
biomol是生物分子;
w是生物分子的亲核基团,优选nh、s或o;
和
x是共价键或c1-c4烷基,
y是亲核基团,优选o、s、nr4、o-nr4、nh-o或nh-nr4,r4为h或c1-c4烷基,
z1、z2、z3各自独立地表示n或c,优选地z3表示c,更优选地z3为c且r3位于z3,
r1为h、经取代或未经取代的c1-c6烷基、经取代或未经取代的链烯基、经取代或未经取代的芳基、或经取代或未经取代的杂环,优选地r1为甲基或乙基,
r2是h或热不稳定和/或酸不稳定的保护基团,
r3是h、经取代或未经取代的c1-c12烷基,例如异丙基、异丁基、仲丁基、叔丁基、异戊基或2,2-二甲基丙基,经取代或未经取代的芳基,经取代或未经取代的杂环,酰基,经取代或未经取代的链烯基,卤素(例如f、cl、br和i)或氰基。
在一个特别实施方案中,biomol选自蛋白质,特别是酶。
在具体优选的实施方案中,biomol是用于核酸聚合反应的酶,例如dna聚合酶。
还公开了用于使由根据本发明的化合物保护的生物分子的亲核基团去保护的方法,所述方法包括分别通过加热和/或酸处理来裂解一个或多个热不稳定和/或酸不稳定基团r2的步骤,以及伴随的生物分子的亲核基团的去保护。
因此,本发明还涉及本发明化合物用于酶的可逆失活的用途。在优选的实施方案中,根据本发明的化合物用于酶的可逆失活,旨在用于核酸聚合反应的酶,例如dna聚合酶。
本发明还涉及用于扩增核酸的方法,其包括使用由根据本发明的化合物灭活的聚合酶,以及在能够裂解一个或多个热不稳定基团r2并使亲核基团去保护的温度下的至少一个热处理步骤,所述热处理步骤之后进行所述核酸扩增步骤。
定义
术语“经取代或未经取代”是指基团中存在的一个或多于一个氢可以被官能团取代,例如选自以下的官能团:胺、亚胺、腈、氰基、酰胺、酰亚胺、羟基、烷氧基、羰基、羧基、酯、硫醇、硫醚、硫酯和卤化物。
术语“cx-cy烷基”是指具有x至y个碳原子的直链或带支链的烷基链或环烷基。直链烷基链的实例包括:甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基和正癸基。带支链的烷基链的实例包括:异丙基、异丁基、仲丁基和叔丁基、异戊基、2,2-二甲基丙基、异辛基、异壬基和异癸基。环烷基的实例包括环戊基、环己基、环庚基和环辛基。
术语“芳基”是指环中不含杂原子的芳族环烃。例如,芳基可以在芳环部分中包含6至14个碳原子。芳基的实例包括:苯基、联苯基、菲基、芘基、草屈基、蒽基和萘基。
术语“杂环”是指包含至少一个饱和或不饱和环的基团,其中至少一个环原子是杂原子,例如n、o或s。杂环可以由多个稠环组成。例如,杂环可以在环部分中包含6至14个原子。
术语“酰基”是指包含羰基的基团,酰基通过羰基的碳原子与分子键合。该碳原子也与属于经取代或未经取代的烷基、经取代或未经取代的芳基或经取代或未经取代的杂环的另一个碳原子键合。例如,酰基包含2至12个碳原子。
术语“链烯基”是指包含至少一个碳-碳双键的、直链或带支链的烷基链或环烷基。链烯基的实例包括:乙烯基、-ch=ch(ch3)、-ch=c(ch3)2、-c(ch3)=ch2、-c(ch3)=ch(ch3)、-c(ch2ch3)=ch2、环己烯基、环戊烯基、环己二烯基、丁二烯基、戊二烯基和己二烯基。例如,链烯基包含2至12个碳原子。
术语“亲核试剂”或“亲核基团”是指能够在合适的反应条件下通过提供形成键所需的两个电子来与反应活性位点(亲电子)形成共价键的基团。一个或更多个亲核基团可以天然存在于生物分子中,例如存在于蛋白质或酶中的赖氨酸的胺基、多肽链的胺末端、丝氨酸的醇、或半胱氨酸的硫醇基团。
术语“保护基团”是指从化学官能团引入分子中以掩蔽其部分或全部反应性的官能团。掩蔽(保护)分子上的化学官能团因而改善了后续反应的选择性。因此,术语“保护”或“经保护的”是指与保护基团反应后的分子的状态。“经保护的生物分子”是具有一个或多于一个经保护基团保护的化学官能团的生物分子。对于酶,经保护的酶于是可以是无活性的。去保护是指从经保护的分子中释放部分或全部保护基团的步骤,并且优选获得在其由本发明的化合物保护之前处于其原始状态的分子。
术语“热不稳定保护基团”是指如下保护基团:所述保护基团在室温下稳定,例如在15℃至25℃,并且通过热处理(加热步骤),例如通过在50℃至100℃的温度下处理而从与其键合的分子裂解、解离或盐析,特别是在适于生物分子(例如酶)的最佳功能的缓冲剂存在下。
热不稳定保护基团尤其包括koukhareva等人的anal.chem.,2009,81,4955-4962;或trinlinkbiotechnologies(wo2012/09434和us8133669)所描述的那些。
热不稳定保护基团的实例通常包括酰胺、醚、酯、缩醛、碳酸酯、硫醚、硫酯、硫代缩醛、硫代碳酸酯,特别是koukhareva等(anal.chem.,2009,81,4955-4962)描述的那些,或单甲氧基三苯甲基(mmt)、二甲氧基三苯甲基(dmt)、和/或经取代或未经取代的苯氧基乙酸酯。
术语“酸不稳定保护基团”是指如下保护基团:其在中性或碱性条件下稳定,并且其在室温的酸性条件下被裂解、解离或盐析,例如,通过在ph低于6.0、甚至低于5.0下热处理而被裂解、解离或盐析。特别地,酸不稳定保护基团的实例包括柠康酰基、叔丁氧基羰基(boc)、苄氧基羰基、三苯甲基(trt)、甲氧基三苯甲基、二甲氧基三苯甲基、苄氧基甲基(bom)或叔丁氧基甲基(bum)。酸不稳定保护基团的其他实例于chem.rev.2009,109(6),第2455-2504页,更具体地第2467和2493页中描述,其取决于它是否为羟基或胺官能团的保护基。
术语“生物分子”广义地理解为包括可由生物有机体合成的所有大分子。特别地,该术语包括核酸聚合物,特别是dna或rna、多糖、肽、多肽和蛋白质,特别是酶。
术语“多肽”、“肽”和“蛋白质”是可互换的,并且是指氨基酸的聚合物或低聚物。这种聚合物的氨基酸通过羧基和两个氨基酸的胺基之间的肽键连接。蛋白质可以包括通过非共价键和/或二硫键连接的多个多肽。
在本发明的含义内,术语“氨基酸”包括天然存在的或非天然存在的氨基酸或其经取代的衍生物。
用于保护生物分子的试剂
本发明化合物或用于保护生物分子的试剂是用于临时保护(“可逆”保护)生物分子(例如蛋白质)的亲核基团(例如胺)的靛红酸酐衍生物。
本发明化合物与生物分子(特别是蛋白质)的亲核基团进行反应。例如,它们可以分别与蛋白质(或酶)赖氨酸的胺ε或蛋白质(或酶)的n-末端胺、蛋白质(或酶)的丝氨酸的醇、或蛋白质的半胱氨酸的硫醇反应以形成酰胺、酯或硫酯键。在蛋白质或酶的亲核官能团中,优选至少一种以必要方式有助于维持蛋白质和/或酶官能团的构象。保护所述必要的亲核官能团(例如赖氨酸、丝氨酸或半胱氨酸)从而导致蛋白质或酶失活。
有利地,由此在生物分子和本发明化合物之间获得的酰胺、酯和/或硫酯键在低温或室温下特别稳定。例如在用于储存或运输的室温条件下,生物分子可以利用本发明的化合物变得失活。更具体地,根据以下原理,可以通过诱导酶的去保护条件来控制酶促反应的起始点:
-在第一步中,通过酶的亲核基团与本发明化合物的酰化反应来使本发明化合物反应,并获得经保护的酶。
-在第二步中,通过酸和/或热处理,通过裂解存在于本发明化合物上并保护本发明化合物的所述亲核官能团的热不稳定和/或酸不稳定的保护基团,使本发明化合物的亲核官能团去保护。
-在第三步中,获得本发明化合物的去保护的亲核官能团与由酶的酰化获得的酰胺、酯或硫酯的羰基之间的环化,其使得同时能够实现酰化逆转、酶去保护和酶活性恢复。
除非在下文中阐明,术语“本发明化合物”是指下式(i)以及所有它们具体描述的子式(特别是式(ii)至(vi))的化合物、这些化合物的盐、它们的立体异构体(包括非对映异构体和对映异构体)、互变异构体和同位素标记的化合物(包括用氘和用放射性同位素取代)。
下式(i)的本发明化合物:
特别地具有:
-氮杂靛红酸酐结构,其使得能够酰化分子的亲核基团,
-亲核官能团y,其经热不稳定和/或酸不稳定基团r2保护,并且一旦释放能够使酰化产生的酰胺键断裂,
-在靛红酸酐和亲核官能团之间的连接臂x,其设计成在亲核官能团脱保护之后促进环化,
-亲核官能团y的热不稳定和/或酸不稳定保护基团r2,其使得能够在加热和/或酸处理步骤期间释放亲核官能团y,从而允许分子内环化,
-优选地,空间大基团r3,其使得能够在亲核试剂周围形成空间位阻和并且加速分子内环化动力学。已经偶然发现,这种空间位阻的存在极大地有利于分子内环化的动力学,从而有利于生物分子的亲核基团的去保护速度。
因此,本发明的目的是下式(i)的化合物:
其中
x是共价键或c1-c4烷基,
y是亲核基团,
z1、z2、z3各自独立地表示n或c,优选z3表示c,更优选z3为c且r3位于z3,
r1为h、经取代或未经取代的c1-c6烷基、经取代或未经取代的烯基、经取代或未经取代的芳基、或经取代或未经取代的杂环,r1优选为甲基或乙基。
r2是热不稳定和/或酸不稳定的保护基团,
r3是h,经取代或未经取代的c1-c12烷基,例如异丙基、异丁基、仲丁基、叔丁基、异戊基或2,2-二甲基丙基,经取代或未经取代的芳基,经取代的或未经取代的杂环,酰基,经取代或未经取代的链烯基,卤素(例如f、cl、br和i)或氰基。
优选地,基团z1、z2和z3中只有一个是n。
在一个具体实施方案中,x是未经取代的c1-c3烷基,例如甲基。
在一个实施方案中,r1是h、c1-c6烷基、链烯基、芳基或杂环,所述烷基、链烯基、芳基或杂环可以由选自腈、氰基、酰胺、酰亚胺、烷氧基、羰基、羧基、酯、硫醚、硫酯和卤化物中一个或多于一个官能团取代。
在一个实施方案中,r1是可以由选自腈、氰基、酰胺、酰亚胺、烷氧基、羰基、羧基、酯、硫醚、硫酯和卤化物的一个或多于一个官能团取代的c1-c6烷基。
在一个实施方案中,r2是选自酰胺、醚、酯、缩醛、碳酸酯、硫醚、硫代酯、硫代缩醛、硫代碳酸酯、单甲氧基三苯甲基(mmt)、二甲氧基三苯甲基(dmt)、任选地被取代的苯氧基乙酸酯、柠檬酸、叔丁氧基羰基(boc))、苄氧基羰基、三苯甲基(trt)、甲氧基三苯甲基、二甲氧基三苯甲基、苄氧基甲基(bom)、或叔丁氧基甲基(bum)的保护基团。
在一个实施方案中,r3是h、c1-c12烷基、芳基、杂环、酰基、链烯基、卤素(例如f、cl、br和i)、或氰基,所述烷基、芳基、杂环基或烯基任选地被选自腈、氰基、酰胺、酰亚胺、烷氧基、羰基、羧基、酯、硫醚、硫酯和卤化物基团的一个或多于一个官能团取代。
在一个实施方案中,r3是c1-c12烷基或卤素(例如f、cl、br和i),所述烷基任选地由选自腈、氰基、酰胺、酰亚胺、烷氧基、羰基、羧基、酯、硫醚、硫酯和卤化物基团的一个或多于一个官能团取代。
r3放置为促进环化动力学。例如,在一个实施方案中,r3优选在位于6处。
在一个具体实施方案中,r3是叔丁基、异丙基、氰基或碘原子。
在一个特定实施方案中,y不同于s-s。在另一个具体实施方案中,亲核二价基团y选自o、s、nr4、o-nr4、nh-o、nh-nr4、c(o)-o-nr4、c(o)-nh-o、c(o)-nh-nr4、o-c(o)-nh-nr4、nh-c(o)-nh-nr4、o-c(o)-nh-o、nh-c(o)-nh-o、o-c(o)-o-nr4、nh-c(o)-o-nr4、c(o)-s,r4为h或c1-c4烷基。
在一个实施方案中,x+y直链原子的数目,即连接环与r2的主链中的原子数(不考虑不同的h或r4取代基)大于或等于2,例如为2至4。
优选地,y选自o、s和nh。在这种情况下,x不是键。
在另一个优选的实施方案中,y选自o、s和nr4,x不是键。在更优选的实施方案中,本发明化合物具有下式(ii):
在一个具体实施方案中,上述式(i)和(ii)中的热不稳定或酸不稳定基团r2选自叔丁氧基羰基(boc)、经取代或未经取代的苯氧基乙酰基、三苯甲基、甲氧基三苯甲基、二甲氧基三苯甲基或柠康酰基。
lebedeva.等人(koukharevai.,lebedeva.,anal.chem.2009,81,4955-4962)已经证实了苯氧基乙酸酯基团的热不稳定性。在一个具体实施方案中,保护基团r2是热不稳定的经取代或未经取代的保护基团苯氧基乙酰基,优选其为由卤素取代的苯氧基乙酰基,例如氟苯氧基乙酰基。
本领域技术人员将能够鉴别特别适合于所需功能的其他热不稳定和/或酸不稳定的保护基团r2,如例如在wo2012/094343、us8,133,669或以下书中描述的:
·peterg.m.wuts的greene'sprotectivegroupsinorganicsynthesis,出版商:wiley-blackwell;版本:第5版(2014年12月23),或
·pjkocienski的protectinggroups,出版商:thiemepublishinggroup;版本:第3次修订版(2005年1月1日)。
在可以与前述实施方案组合的根据式(ii)化合物的另一具体实施方案中,r1为甲基。
在可以与前述实施方案组合的根据式(ii)化合物的另一具体实施方案中,r3是卤素,例如碘。
在可以与前述实施方案组合的根据式(ii)化合物的另一具体实施方案中,z1为n且z2为c。
特别地,本发明涉及具有以下结构的化合物之一,其也在实施例中进行了描述:
用于制备本发明化合物的方法
根据实施例中描述的方法或现有技术中已知的其他合成方法,可以容易地合成本发明的化合物。
例如,根据式(ii)的本发明的一些化合物可以由前体羟基化化合物6合成,其合成于实施例1中描述。然后可以通过使热不稳定或酸不稳定基团r2与前体的羟基官能团反应来改性前体化合物6。还可以通过常规化学方法使r1和/或r3基团与这些前体化合物或其衍生物反应。根据式(ii)的本发明化合物最终可以通过亲核取代的环化获得,例如如下面实施例1中的图3的步骤(i)所述,以得到本发明化合物。
保护生物分子的方法
本发明的化合物可用于酶、蛋白质、以及更一般的包含一个或多于一个亲核基团的生物分子的保护、可逆修饰或失活,例如包含一种或多于一种胺的生物分子。
本发明特别涉及一种用于制备包含一个或多于一个经保护的亲核基团的生物分子的方法,其包括将如上所述的本发明化合物与生物分子接触,以形成包含一个或多于一个经保护的亲核基团的生物分子(下文也称为“经保护的生物分子”),其条件是能够酰化所述生物分子的一个或多于一个亲核基团。
通过用生物分子的亲核基团的加入对酸酐的一个羰基官能团进行亲核取代,并消除co2来进行酰化。
在一个特别优选的实施方案中,生物分子包含胺官能团。特别地,生物分子是包含赖氨酸残基的ε-胺官能团或末端氨基酸的胺的蛋白质。在这些实施方案中,赖氨酸残基的ε-胺官能团或末端胺的酰化使得能够形成非常稳定的酰胺键,特别是在长期储存条件下(例如在室温下超过24小时或甚至数天)如此。
作为举例说明,酰化反应如下图1所示,其使用优选的式(ii)试剂,其中r1是甲基,z1是n,z2是c,r3是碘原子。
方案1:本发明化合物对生物分子的酰化反应
在其中生物分子包含多个待保护的亲核基团的情况下,可以根据所需的修饰(保护)程度来调节所用试剂的量。特别地,在酶的情况下,优选所需的修饰程度对应于修饰阈值;若超过该修饰阈值,则酶失活。该阈值可以通过简单测试凭经验来确定。
用于保护反应的缓冲液通常是ph为6至9、优选7至9、更优选7至8的缓冲液。缓冲液的实例包括磷酸盐缓冲液和在该ph范围中操作的非亲核缓冲液,其能够含有高达50%的dmso。这些缓冲液是技术人员已知的,如在http://www.sigmaaldrich.com/life-science/core-bioreagents/biological-buffers.html中所示的。
酰化反应,特别是酶与至少一种式(i)或(ii)化合物的酰化反应优选在磷酸盐缓冲液中、例如在ph7.4下获得。
保护过程优选地在4℃至40℃的温度下进行,例如在4℃至25℃的温度下进行。
包含能够通过上述方法获得的一个或多于一个经保护的亲核基团的生物分子也是本发明的一部分。特别地,由于其多肽序列中存在多个赖氨酸,酶可以包含多个受保护的胺基团,或者由于丝氨酸或半胱氨酸的存在,酶可以分别包含oh或sh基团。
因此,本发明涉及下式(iii)的经保护的生物分子:
其中
biomol是生物分子,
w是生物分子的亲核基团,优选nh、s或o,
x是共价键或c1-c4烷基,
y是亲核基团、o、s、nr4、o-nr4、nh-o或nh-nr4,r4是h或c1-c4烷基,
z1、z2、z3各自独立地表示n或c,优选z3表示c,更优选z3为c且r3位于z3位置,
r1为h,经取代或未经取代的c1-c6烷基,经取代或未经取代的链烯基,经取代或未经取代的芳基,或经取代或未经取代的杂环,r1优选为甲基或乙基,
r2是h或热不稳定和/或酸不稳定保护基团,
r3是h、经取代或未经取代的c1-c12烷基,例如异丙基、异丁基、仲丁基、叔丁基、异戊基或2,2-二甲基丙基,经取代或未经取代的芳基,经取代或未经取代的杂环,酰基,经取代或未经取代的链烯基,卤素(例如f、cl、br和i)或氰基。
当然,当生物分子包含多个亲核基团,例如包含几个赖氨酸的蛋白质时,单个生物分子可包含部分或全部这些亲核基团,因此通过用本发明的试剂酰化来保护。
特别地,当式(ii)的试剂与包含一个或多于一个亲核基团的生物分子反应时,获得下式(vii)的经保护的生物分子:
通过选择上述式(iv)、(v)和(vi)之一的试剂,分别获得根据式(viii)、(ix)和(x)之一的经保护的生物分子。
在一个具体实施方案中,生物分子(或biomol)选自酶。
该方法适用于任何类型的酶。感兴趣的特定酶包括分子生物学技术中用于基因工程或核酸聚合中使用的酶,更具体为体外诊断技术中使用的酶。
作为举例说明,这些酶包括脂肪酶、蛋白酶、糖苷酶或核酸酶。
特别地,感兴趣的酶包括限制酶,连接酶,rna聚合酶,dna聚合酶如dna聚合酶i、ii或iii,或dna聚合酶α、β或γ,末端脱氧核苷酸转移酶(tdt)或端粒酶,dna-依赖性rna聚合酶,引物,或dna-依赖性rna聚合酶(逆转录酶)。
在更特别优选的实施方案中,根据本发明的生物分子是旨在用于核酸聚合反应的酶,例如dna聚合酶。适用于核酸聚合的聚合酶是本领域技术人员熟知的。聚合反应具体是在聚合酶链反应(pcr)扩增过程中的聚合反应。
在一个实施方案中,在根据本发明的保护方法中使用的感兴趣的酶选自以下聚合酶:t7dna聚合酶、kornbergdna聚合酶、klenowdna聚合酶、taqdna聚合酶、micrococcaldna聚合酶、αdna聚合酶、pfudna聚合酶、amv逆转录酶、mmlv逆转录酶、大肠杆菌rna聚合酶、sp6、t3或t7rna聚合酶。特别地,在一个实施方案中,在根据本发明的方法中使用热稳定和/或具有核酸外切酶活性的dna聚合酶(例如3'5'核酸外切酶)来制备经保护的聚合酶。
这些经保护的聚合酶特别适用于热启动应用。保护聚合酶防止了dna在低温下非特异性扩增,从而提高扩增反应的效率,其中所述酶仅在高温下通过切割热不稳定基团来去保护。
在这些热稳定聚合酶中,我们将特别列出在50℃至100℃之间不易于发生结构变性和/或酶活性失活的那些、在一旦去保护后在50℃至100℃之间起作用并活化的那些。具体地,聚合酶:taq聚合酶和klentaq聚合酶。
其他热稳定酶可以来自以下生物有机体:thermusantranikianii、thermusaquaticus、thermuscaldophilus、thermuschliarophilus、thermusfiliformis、thermusflavus、thermusigniterrae、thermuslacteus、thermusoshimai、thermusruber、thermusrubens、thermusscotoductus、thermussilvanus、thermusspeciesz05、thermusspeciessps-17、thermusthermophilus、thermotogamaritima、thermotoganeapolitana、thermosiphoafricanus、anaerocellumthermophilum、bacilluscaldotenax、bacillusstearothermophilus等。
使用本发明化合物的方法
根据所选择的r2基团,根据本发明保护的生物分子可以有利地通过简单的加热和/或酸处理来脱保护。
通过加热或酸处理使根据式(vii)的受保护的生物分子(酶)去保护的原理在下图2中描述,其中r1是甲基,z1是n,z2是c,并且r3是碘原子:
方案2:使根据本发明的经保护的生物分子去保护的实例方法
加热或酸处理通过裂解热不稳定或酸不稳定基团来释放本发明化合物的亲核官能团。然后,如此释放的亲核官能团可以通过生物分子的环化和去保护而逆转酰化。有利地,去保护反应产生的副产物是非反应性的。此外,该保护反应不需要使用tris缓冲液或产生酸性和高温条件的缓冲液。特别地,去保护步骤可以通过在ph为6.5至9.5、优选8至9.0、更优选8.5至9.0的条件下进行热处理来进行。
因此,还描述了用于使被本发明化合物保护的生物分子的亲核基团去保护的方法,所述方法包括分别通过热和/或酸处理使热不稳定和/或酸不稳定基团r2裂解和伴随的亲核基团去保护的步骤。
上述方法的一个优点是其能够使酶失活。“失活”应理解为,与保护前最佳活性条件下的催化活性相比,通过本发明方法保护的酶的催化活性大大降低或甚至不存在。
然后,上述去保护方法能够恢复经保护的酶的酶活性。
因此,本发明还涉及本发明化合物用于酶的可逆失活的用途。在一个优选的实施方案中,本发明化合物用于使旨在用于核酸扩增反应的酶如聚合酶可逆地失活。
本发明特别适用于酶热启动应用,例如用于核酸扩增反应(称为“pcr”的反应)的聚合酶。
因此,本发明还涉及扩增核酸的方法,其包括:(i)使用由本发明化合物保护或灭活的聚合酶,(ii)用于使聚合酶去保护的至少一个步骤,例如,通过在使得热不稳定基团r2裂解的温度下进行热处理,(iii)使用步骤(ii)中去保护的聚合酶进行的核酸扩增步骤。
在一个具体实施方案中,使用dna聚合酶如taq聚合酶、pfu聚合酶、t7聚合酶、来自大肠杆菌dna聚合酶的klenow片段和/或逆转录酶,或如上文所述的任何其他聚合酶来实施扩增方法。
在可以与上述组合的另一实施方案中,扩增方法是本领域技术人员熟知的聚合酶链式反应(pcr反应)。pcr方案包括20至40个循环,例如,每个循环至少包括:(i)在通常为90℃至95℃的温度下待扩增的dna的变性阶段,(ii)在通常为55℃至65℃的温度下引物与待扩增的dna杂交的阶段,和(iii)在通常为68℃至75℃的温度下的延伸阶段。
在这种情况下,根据本发明的经保护聚合酶优选是经保护的热稳定聚合酶,例如taq(栖热水生菌,thermusaquaticus)保护的聚合酶、pfu(强烈火球菌,pyrococcusfuriosus)保护的聚合酶、vent或tli(嗜热高温球菌,thermococcuslitoralis)保护的聚合酶,或其变体,特别是重组体。优选地,保护所述聚合酶的足够数量的胺,使得经保护的聚合酶在室温下无活性或基本上无活性,并且可以在等于或高于用于pcr扩增的引物退火温度的温度下去保护。
本发明的经保护酶和聚合酶也有利地应用于通过pcr进行核酸扩增的变体方法中。我们特别列出巢式pcr、定量pcr(或qpcr)、半定量或实时pcr、“易错”pcr、或逆转录pcr(rt-pcr)
试剂盒和工具包
本发明还涉及试剂盒或工具包,其包括根据本发明的经保护酶以及任选的试剂、缓冲剂、对照物和/或说明书。
特别地,本发明涉及用于热启动核酸扩增的试剂盒,其包括:
i.根据本发明保护的热稳定dna聚合酶,例如式(iii)、(vii)、(viii)、(ix)或(x),其中biomol是热稳定dna聚合酶,
ii.在适当的情况下,核酸检测试剂,例如荧光试剂,
iii.在适当的情况下,缓冲剂、dntp等。
这些试剂盒特别适用于实施如上所述的扩增方法。
借助于下面详述的实施例和附图,将更好地理解本发明。
附图说明
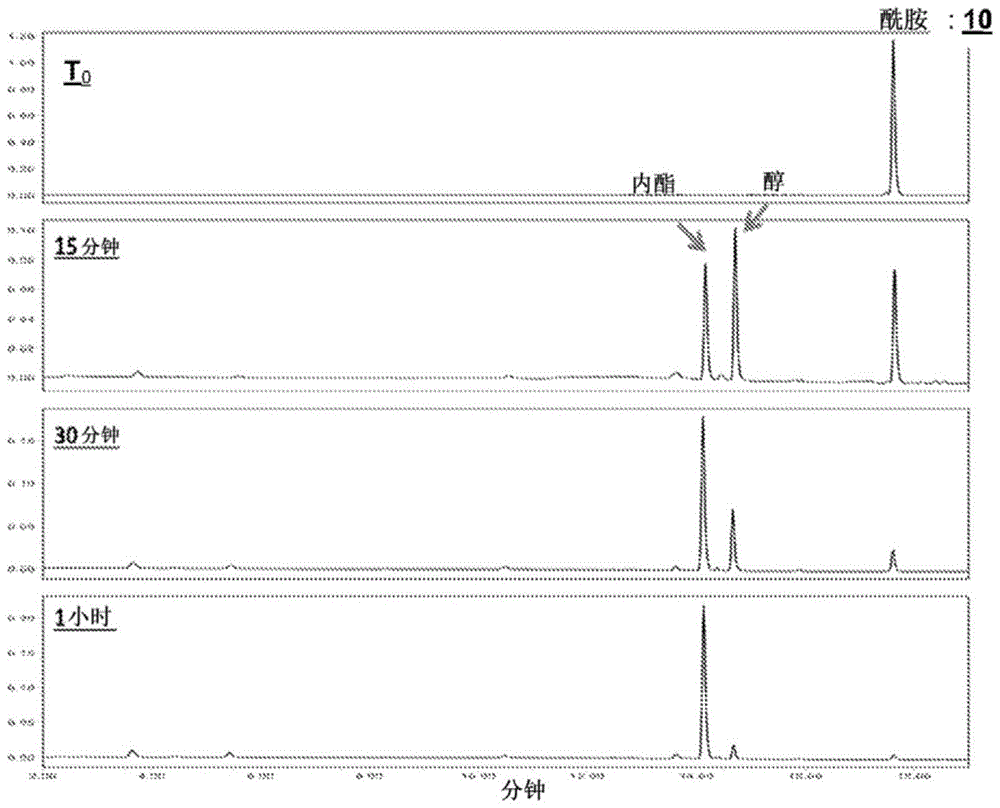
图1:苯氧基乙酸氮杂-邻氨基苯甲酸酯衍生物10的裂解和苯乙胺的释放的hplc监测。
图2:氟代苯氧基乙酸酯氮杂-邻氨基苯甲酸酯衍生物14的裂解和苯乙胺的释放的hplc监测。
图3:n-boc氮杂-邻氨基苯甲酸酯衍生物19的裂解和苯乙胺的释放的hplc监测。
图4:由不同浓度的化合物13酰化血红蛋白。
图5:热处理后血红蛋白酰化的逆转。
上图(a):8mguhc中的血红蛋白
中图(b):由化合物13酰化然后加入8mguhcl中的血红蛋白
下图(c):由化合物13酰化然后在trisph9中加热至95℃并加入8mguhcl中的血红蛋白。
图6:由化合物13酰化之后taq聚合酶失活,然后在pcr条件下在95℃热处理15分钟后恢复其活性(实验进行两次,显示平均值)。左侧带点的白色:没有活化;右侧黑色交叉阴影:在95℃下活化15分钟后。
实施例
在下面描述的示例中,使用以下缩写:
·acn:乙腈,
·acoet:乙酸乙酯,
·boc2o:二碳酸二叔丁酯,
·tlc:薄层色谱,
·cdcl3:氘代氯仿
·d:双峰,
·dcm:二氯甲烷
·dd:双二重峰,
·dmf:二甲基甲酰胺
·dmso:二甲基亚砜,
·dmso-d6:氘代二甲基亚砜,
·milliq水:超纯水(millipore,molsheim,france)
·edc:n-乙基-n’-(3-二甲基氨基丙基)碳二亚胺
·eq:当量,
·pe:石油醚,
·et2o:乙醚,
·hplc:高效液相色谱,
·lcms:与质谱仪耦合的液相色谱仪
·hobt:羟基苯并三唑,
·ia:靛红酸酐,
·m:多重峰,
·nd:未测定
·nis:n-碘代琥珀酰亚胺
·q:四重峰,
·yld:收率,
·rf或rt:保留时间,
·nmr:核磁共振,
·s:单峰
·t:三重峰,
·rt:室温
·tbdms:叔丁基二甲基甲硅烷基
·tea:三乙胺
·thf:四氢呋喃
一般条件
以下实施例中使用的化合物的分析和合成的一般条件如下:
使用配备有pda996二极管阵列检测器(waters)、zq2000质谱检测器(waters)、empower软件版本2和在30℃下以1ml/分钟的流速使用的watersxterramsc18色谱柱(4.6×30,2.5μm)的waters2795alliancehplc系统进行hplc分析(在260nm处检测或最大图)。zq2000质谱仪具有电喷雾电离源。电离以正态模式进行,锥电压为20v,毛细管电压为3.5kv。
用于hplc分析的条件是:
在jeollambda400mhz光谱仪上记录nmr光谱。化学位移(δ)相对于用作内部参考的溶剂的峰值以ppm表示(cdc13:7.26ppm;dmso-d6:2.49ppm)。使用上述缩写来描述光谱:s、d、t、q、qu和m。耦合常数(j)以赫兹(hz)表示。
柱色谱在macherey-nagelkieselgel60、筛网0.063-0.2mm/70-230或merck
通过薄层色谱分析在macherey-nagel
实施例1:具有o-苯氧基乙酸酯热不稳定保护基团(9)的本发明的氮杂靛红酸酐分子的合成(对应于化合物iv)
在该实施例中描述了本发明化合物的合成(根据方案3)。首先在六个步骤中合成初始的前体羟基化化合物6。它可以作为合成本发明其他化合物的起始化合物。然后将化合物6进行苯氧基乙酰化,得到化合物7,然后碘化,得到化合物8,最后环化得到期望的氮杂靛红化合物9,其能够与蛋白质或其他生物分子反应以临时保护它。
方案3:邻苯氧基乙酰基化合物9的合成。a)litmp,thf,3h,-50℃;dmf,1小时30分钟,-80℃,rt;hc1ph2;b)net3,dcm,10分钟,rt;t-bubr,ag2o,2小时,rt;c)nabh4,etoh,30分钟,-10℃;d)tbdmscl,咪唑,dcm,4小时,rt;e)menh2,h2o,t-buoh,24小时,100℃;f)tbaf,acoh,dcm,15小时,rt;g)苯氧基乙酰氯,hobt,dcm,5min,rt然后6,24小时,rt;h)nis,dcm,acoh,2-3小时,rt.i)cocl2,tea,et2o,30分钟,rt。
实施例1.1:4-氯-1-羟基呋喃[3,4-c]吡啶-3(1h)-酮(1)的合成
在氮气下的500mlshlenk管中,将19.30ml的2,2,6,6-四甲基哌啶(114.24mmol,3当量)溶解在150mlthf中。在-10℃下,加入60.13ml的n-buli(1.9m于己烷溶液中,114.24mmol,3当量),并将反应混合物搅拌10分钟。在-80℃下,加入6.00g的2-氯烟酸(38.08mmol,1当量),并将反应在-50℃下搅拌3小时。然后在-80℃下引入17.69ml的dmf(228.48mmol,6eq)并将反应混合物搅拌1小时30分钟。恢复到室温后,加入100ml水,用acoet萃取溶液(3×150ml)。然后用浓hcl溶液将水相酸化至ph2,然后用acoet萃取(3×200ml)。然后合并有机相,用mgso4干燥,过滤,最后蒸发。然后使用梯度洗脱(dcm至dcm/acoet85/15)在硅胶柱上纯化得到的油。
得到最终产物为白色粉末,产率69%(4.88g,26.30mmol)。
mp=191-193℃;ir(kbr):v,3100(oh),2913,2766,1776(c=o),1609,1584,1408,1194,1136,1093,1047,925,755cm-1;1hnmr(400mhz,dmso-d6):δ6.67(bs,1h);7.79(d,1h,3j=5.0hz);8.50(bs,1h);8.74(d,1h,3j=5.0hz);13cnmr(100mhz,dmso-d6):δ96.9;118.8;120.4;147.0;154.6;159.3;164.6。
实施例1.2:2-氯-4-甲酰基烟酸叔丁酯(2)的合成
在室温下向250ml烧瓶中加入3.50g的1(18.86mmol,1当量)、35ml的dcm和2.62ml的tea(18.86mmol,1当量)。将混合物在室温下搅拌10分钟。然后在0℃下分批加入8.54ml的t-bubr(75.45mmol,4当量)和8.74g的ag2o(37.72mmol,2当量)。将反应在室温下搅拌2小时。将混合物通过硅藻土过滤,蒸发,并在硅胶柱上使用梯度洗脱(环己烷/acoet95/5至环己烷/acoet90/10)纯化。得到最终产物为白色粉末,产率67%(3.07g,12.70mmol)。
mp=97-99℃;ir(kbr):v,3077,1732(c=o),1708(c=o),1578,1370,1294,1261,1178,1133,847cm-1;1hnmr(400mhz,cdcl3):δ1.64(s,9h);7.65(d,1h,3j=5.0hz);8.65(d,1h,3j=5.0hz);10.06(s,1h);13cnmr(100mhz,cdcl3):δ27.9(3c),85.1;121.0;130.1;140.6;149.1;150.9;163.0;188.2。
实施例1.3:2-氯-4-(羟甲基)烟酸叔丁酯(3)的合成。
在100ml烧瓶中,将3.00g的2(12.41mmol,1当量)溶于35ml的etoh中。在-10℃下,分批加入0.51g的nabh4(13.65mmol,1.1当量)并将反应混合物搅拌30分钟。然后加入50ml水,用dcm(3×75ml)萃取溶液。然后合并有机相,用饱和nacl溶液(2×50ml)洗涤,经mgso4干燥并蒸发。
得到最终产物为白色粉末,产率97%(2.94g,12.06mmol)。
mp=122-124℃;ir(kbr):v,3263(oh),3003,2980,1717(c=o),1592,1387,1365,1299,1170,1131,1080,1063,869,846cm-1;1hnmr(400mhz,cdcl3):δ1.60(s,9h);2.89(t,1h,3j=6.3hz);4.69(d,2h,3j=6.3hz);7.39(d,1h,3j=5.0hz);8.37(d,1h,3j=5.0hz);13cnmr(100mhz,cdcl3):δ27.9(3c);61.3;84.3;120.6;128.6;147.2;149.8;150.8;164.8。
实施例1.4:4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-2-氯烟酸叔丁酯(4)的合成
在室温下向100ml烧瓶中加入2.25g的3(9.23mmol,1当量)、30ml的dcm、1.88g的咪唑(27.70mmol,3当量)和2.78g的tbdmscl(18.47mmol,2当量)。将反应混合物在室温下搅拌4小时。然后加入50ml水,并用dcm(3×75ml)萃取溶液。然后合并有机相,用饱和nacl溶液(2×50ml)洗涤,经mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/et2o95/5)在硅胶柱上纯化粗反应产物。得到最终产物为无色油状物,产率为95%(3.15g,8.80mmol)。
ir(kbr):v,2956,2931,2359,1717(c=o),1584,1369,1291,1259,1168,1127,840cm-1;1hnmr(400mhz,cdcl3):δ0.10(s,6h);0.94(s,9h),1.60(s,9h);4.74(s,2h);7.50(d,1h,3j=5.0hz);8.39(d,1h,3j=5.0hz);13cnmr(100mhz,cdcl3):δ-5.6(2c);18.2;25.7(3c),27.9(3c);61.1;83.6;119.7;127.6;146.7;149.7;151.0;164.1。
实施例1.5∶4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-2-甲基氨基)烟酸叔丁酯(5)的合成
在室温下向密封管中加入4.00g的4(11.17mmol,1当量)、15ml的t-buoh和4.83ml的menh2(40%重量/重量于h2o中,55.87mmol,5当量)。然后将反应混合物在100℃下搅拌24小时。蒸发后,然后加入50ml水,并用dcm(3×75ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/et2o90/10)在硅胶柱上纯化粗反应产物。
得到最终产物为无色油状物,产率79%(3.10g,8.79mmol)。
ir(kbr):v,3377(n-h),2931,2857,1732(c=o),1584,1370,1291,1259,1190,1169,1127,1112,839cm-1;1hnmr(400mhz,cdcl3):δ0.11(s,6h);0.96(s,9h);1.59(s,9h);3.02(d,3h,3j=4.9hz);4.90(s,2h);6.97(d,1h,3j=5.2hz);7.87(bs,1h);8.25(d,1h,3j=5.2hz);13cnmr(100mhz,cdcl3):δ-5.4(2c);18.4;25.9(4c),28.4(3c);63.9;82.2;104.9;109.0;151.8;154.6;159.5;167.8。
实施例1.6:4-(羟甲基)-2-(甲氨基)烟酸叔丁酯(6)的合成
在100ml烧瓶中,将3.00g的5(8.51mmol,1当量)溶解在30ml的dcm中。同时加入0.97ml的acoh(17.02mmol,2当量)和17.02ml的tbaf(1mthf溶液,17.02mmol,2当量)。将反应在室温下搅拌15小时。然后加入70ml水,并用dcm(3×70ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet80/20至pe/acoet60/40)在硅胶柱上纯化粗反应产物。
得到最终产物为白色粉末,产率92%(1.86g,7.81mmol)。
ir(kbr):v,3348(n-h),3242(o-h),2980,2944,1667(c=o),1596,1557,1530,1367,1242,1165,1126,1074,1061,805cm-1;1hnmr(400mhz,cdcl3):δ1.60(s,9h),3.01(d.3h,3j=4.9hz);4.73(s,2h);6.73(d,1h,3j=5.2hz);7.58(bs,1h);8.22(d,1h,3j=5.2hz);13cnmr(100mhz,cdcl3):δ28.3(3c);28.4;64.0;83.0;106.4;110.7;151.8;153.2;159.2;167.5。
实施例1.7:2-(甲基氨基)-4-((2-苯氧基乙酰氧基)甲基)烟酸叔丁酯(7)
在50ml烧瓶中,将204μl苯氧基乙酰氯(1.47的mmol,1当量)溶于5mldcm中。加入198mg的hobt(1.47mmol,1当量),并将反应混合物在室温下搅拌。10分钟后,加入350mg的6(1.47mmol,1当量),然后将反应在室温下搅拌24小时。然后加入40ml水,并用et2o(4×30ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/acoet8/2)在硅胶柱上纯化粗反应产物。
得到最终产物为白色粉末,产率为70%(385mg,1.03mmol)。
mp=149-151℃;ir(kbr):v,3365,2970,1760,1668.1583,1192,752;1hnmr(cdcl3,400mhz):δ1.59(s,9h);3.01(d,3h,3j=4.9hz);4.75(s,2h);5.45(s,2h);6.53(d,1h,3j=5.1hz);6.94(m,2h);7.01(t,1h,3j=7.3hz);7.30(m,2h);7.89(d,1h,3j=4.2hz);8.18(d1h,3j=5.12hz);13cnmr(cdcl3,100mhz):δ28.3(4c);65.2;65.4;83.1;105.6;108.9;114.6(2c);121.8;129.6(2c);147.4;152.0;157.6;159.6;167.1;168.6。
实施例1.8:5-碘-2-(甲基氨基)-4-(((2-苯氧基乙酰氧基)甲基)烟酸叔丁酯(8)
向25ml烧瓶中加入280mg的7(0.75mmol,1当量)、5ml的dcm、253mg的nis(1.12mmol,1.5当量)和500μl乙酸。将反应混合物在室温下搅拌3小时。然后将反应混合物用20ml饱和硫代硫酸钠水溶液中和,加入20ml饱和nahco3溶液,然后用et2o(4×30ml)萃取。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/acoet9/1)在硅胶柱上纯化粗反应产物。
得到最终产物为黄色粉末,产率91%(340mg,0.68mmol)。
mp=127-129℃;ir(kbr)v,3424,2934,1755,1704,1576,1193,752;1hnmr(cdcl3,400mhz):δ1.56(s,9h);2.97(d,3h,3j=4.6hz);4.64(s,2h);5.4(s,2h);6.90(d,2h,3j=7.8hz);6.99(t,1h,3j=7.3hz);7.27(m,2h);8.50(s,1h);n-h信号消失;13cnmr(cdcl3,100mhz):δ28.1(3c);28.4;65.0;68.7;82.5;83.7;112.7;114.6(2c);121.8;129.5(2c);144.6;157.5;157.7;158.3;166.5;168.3。
实施例1.9:(6-碘-1-甲基-2,4-二氧代-2,4-二氢-1h-吡啶并[2,3-d][1,3]口恶嗪-5-基)甲基2-苯氧基乙酸酯(9)
在氮气下的50ml烧瓶中,将300mg的8(0.60mmol,1当量)溶于10ml的dcm中。同时加入951μl光气(20%于甲苯溶液中,1.80mmol,3当量)和251μl的tea(1.80mmol,3当量),并将反应在室温下搅拌。重复三次以转换所有原料。然后加入40ml水,并用et2o(5×50ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。将反应混合物蒸发至干燥,并使用梯度洗脱(h2o/acn95/5至h2o/acn5/95)在c18-接枝的硅胶柱上直接纯化。得到最终产物为白色粉末,产率为90%(255mg,0.54mmol)。
mp=162-164℃;ir(kbr)v(cm-1):3439,2935,1787,1757,1728,1450,1176,756;1hnmr(cdcl3,400mhz):δ3.65(s,3h);4.65(s,2h);5.79(s,2h);6.86(d,2h,3j=8.5hz);6.96(t,1h,3j=7.5hz);7.27(m,2h);9.00(s,1h);13cnmr(cdcl3,100mhz):δ31.2;64.8;66.7;94.7;107.7;114.6(2c);121.8;129.5(2c);146.8;150.0;153.3;155.1;157.5;162.7;168.2。
实施例2:具有o-氟-苯氧基乙酸酯热不稳定保护基团13的本发明的一类氮杂靛红酸酐分子(对应于化合物v)的合成
此处描述的是本发明的另一种示例性化合物的合成(根据方案4)。前体化合物6是氟-苯氧基乙酰基(可以是对热更不稳定的基团),以得到化合物11,然后碘化得到化合物12,最后环化得到氮杂异构化合物13。
方案4:o-氟-苯氧基乙酰基化合物13的合成。g)x=f,4-氟苯氧基乙酸,edc,hobt,5分钟,rt,然后6,3小时,rt;h)nis,dcm,acoh,2小时至3小时,rt;i)cocl2,tea,et2o,30分钟,rt.。
实施例2.1:4-((2-(4-氟苯氧基)乙酰氧基)甲基)-2-(甲基氨基)烟酸叔丁酯(11)
向50ml烧瓶中加入149mg的4-氟苯氧基乙酸(0.88mmol,1.05当量)、5ml的dcm、169mg的edc(0.88mmol,1.05当量)和119mg的hobt(0.88mmol,1.05当量)。在室温下5分钟后,加入200mg的6(0.84mmol,1当量),并将反应在室温下搅拌3小时。然后加入30ml水,并用et2o(3×30ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet9/1至pe/acoet8/2)在硅胶柱上纯化粗反应产物。
得到最终产物,为淡黄色粉末,产率57%(190mg,0.49mmol)。
mp=151-153℃;ir(kbr)v,(cm-1):3376,2977,2935,1758(co),1683(co),1586,1189,831;1hnmr(cdcl3,400mhz):δ1.60(s,9h);3.02(d,3h,3j=4.6hz);4.71(s,2h);5.45(s,2h);6.54(d,1h,3j=5.2hz);6.88(m,2h);6.99(t,2h,3j=8.3hz);7.88(s,1h);8.20(d,1h,3j=5.2hz);13cnmr(cdcl3,100mhz):δ28.1;28.3(3c);65.4;65.9;83.1;105.6;109.0;115.9(d,3jc-f=8hz,2c);116.0(d,2jc-f=23hz,2c);147.3;152.0;153.8(d,4jc-f=2hz,1c);157.8(d,1jc-f=239hz,1c);159.5;167.1;168.4。
实施例2.2:(6-碘-1-甲基-2,4-二氧代-2,4-二氢-1h-吡啶并[2,3-d][1,3]口恶嗪-5-基)甲基-2-(4-氟苯氧基)乙酸酯(12)
在25ml烧瓶中,将200mg的11(0.51mmol,1当量)溶于3ml的dcm中。加入172mg的nis(0.77mmol,1.5当量)和150μl乙酸,将反应介质搅拌2小时。然后将反应混合物用20ml饱和硫代硫酸钠水溶液中和,加入20ml饱和nahco3溶液中,然后用et2o(4×30ml)萃取。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/acoet9/1)在硅胶柱上纯化粗反应产物。得到最终产物为黄色油状物,产率68%(180mg,0.35mmol)。
ir(kbr):v,3413,2926,1763,168,8,1506.1183,828;1hnmr(cdc13,400mhz);δ1.56(s,9h);2.97(d,3h,3j=4.64hz);4.60(s,2h);5.37(s,2h);6.85(m,2h);6.96(m,2h);8.50(s,1h);13cnmr(cdcl3,100mhz):δ28.1(3c);28.4;65.7;68.7;82.4;83.7;112.7;115.9(d,2jc-f=23hz,2c);116.0(d,3jc-f=8hz,2c);144.5;153.7;157.7;157.8(d,1jc-f=239hz,1c);158.3;166.4;168.2。
实施例2.3:(6-碘-1-甲基-2,4-二氧代-2,4-二氢-1h-吡啶并[2,3-d][1,3]口恶嗪-5-基)甲基-2-(4-氟苯氧基)乙酸酯(13)
在氮气下的50ml烧瓶中,将160mg的12(0.31mmol,1当量)溶于3mldcm中。同时加入489μl光气(20%于thf溶液中,0.98mmol,9当量)和129μl的tea(0.98mmol,9当量),并将反应搅拌30分钟。然后加入30ml水,用et2o(3×30ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。将反应混合物蒸发至干,并使用梯度洗脱(h2o/acn95/5至h2o/acn5/95)在c18-接枝的硅胶柱上直接纯化。
得到最终产物为白色粉末,产率为77%(115mg;0.24mmol)。
mp=150-152℃;ir(kbr)v(cm-1):3454,3078,2934,1787,1745,1504,1194,825;1hnmr(cdcl3,400mhz):δ3.66(s,3h);4.62(s,2h);5.78(s.2h);6.83(m,2h);6.95(m,2h);9.01(s,1h);13cnmr(cdcl3,100mhz):δ31.2;65.6;66.8;94.7;107.7;115.9(d,2jc-f=23hz,2c);115.9(d.3jc-f=8hz,2c);146.7;150.0;153.4;153.7(d,4jc-f=2hz,1c);155.2;157.8(d,1jc-f=239hz,1c);162.8;168.1。
实施例3:具有n-boc酸不稳定/热不稳定保护基团18的本发明的一类氮杂靛红酸酐分子(对应于化合物vi)的合成
在此处,我们描述了本发明另一种示例性化合物的合成(根据方案5)。首先将前体化合物6转化为叠氮化合物15,其为胺化合物的前体,其中胺官能团与boc保护基团偶联,得到化合物16。碘化反应得到化合物17,最后环化得到氮杂靛红酸酐化合物18。
方案5:n-boc化合物18的合成。k)meso2cl,net3,dcm,3小时,0℃,rt然后nan3,tea,dcm,3小时,rt.;l)boc2o,naoh,pcy3,thf,2小时,rt.;m)nis,dcm,acoh,12小时,rt.;n)cocl2,tea,et2o,30分钟,rt.
实施例3:4-(叠氮甲基)-2-(甲基氨基)烟酸叔丁酯(15)
在氮气下的100ml烧瓶中,将1.40g的6(5.88mmol,1当量)溶于20ml的dmf中。在0℃下,同时加入0.91ml的meso2cl(11.76mmol,2当量)和4.08ml的tea(29.3mmol,5当量)。将反应混合物在室温下搅拌并通过tlc(pe/et2o1/1)进行监测。在全部形成相应的甲磺酸酯衍生物后,加入1.15g的nan3(17.64mmol,3当量),并将混合物在室温下搅拌3小时。然后加入50ml水,并用et2o(3×60ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/et2o9/1至pe/et2o8/2)在硅胶柱上纯化粗反应产物。得到最终产物为黄色油状物,收率73%(1.13g,4.29mmol)。
ir(kbr)v,3374,2978,2104(n3),1679,1586,1425,1125,847,655;1hnmr(cdc13,400mhz):δ1.60(s,9h);3.01(d,3h,3j=5.0hz);4.58(s,2h);6.61(d,1h,3j=5.0hz);7.73(s,1h);8.24(d,1h,3j=5.0hz);13cnmr(cdcl3,100mhz):δ28.3(3c);53.9;77.0;83.1;106.7;111.5;146.8;152.0;159.5;167.1。
实施例3.1:4-(((叔丁氧基羰基)氨基)甲基)-2-(甲基氨基)烟酸叔丁酯(16)
在25ml烧瓶中,将180mg的15(0.68mmol,1当量)和447mg的boc2o(2.05mmol,3当量)溶解在5mlthf中。依次加入750μlnaoh水溶液(0.75mmol,1.1当量,1m于h2o中)和230mg的pcy3。将反应在室温下搅拌2小时。然后加入30ml水,并用et2o(3×30ml)萃取溶液。然后合并有机相,并用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet9/1至pe/acoet8/2)在硅胶柱上纯化粗反应产物。得到最终产物为黄色油状物,产率59%(135mg,0.40mmol)。
1hnmr(cdcl3,400mhz):δ1.44(s,9h);1.60(s,9h);3.01(d,3h,3j=4.6hz);4.40(d,2h,3j=6.1hz);5.02(sl,1h);6.57(d,1h,3j=5.1hz);7.52(sl,1h);8.18(d,1h,3j=5.1hz);13cnmr(cdcl3,100mhz):δ28.4(6c);28.5;43.9;79.6;83.0;107.6;111.6;150.5;151.5;155.7;159.2;167.4。
实施例3.2:4-(((叔丁氧基羰基)氨基)甲基)-5-碘-2-(甲基氨基)烟酸叔丁酯(17)
在25ml烧瓶中,将130mg的16(0.39mmol,1当量)溶于4ml的dcm中。加入225mg的nis(0.58mmol,1.5当量)和200μl乙酸,将反应混合物搅拌12小时。然后将反应混合物用10ml饱和硫代硫酸钠水溶液中和,加入10ml饱和nahco3溶液中,然后用et2o(4×30ml)萃取。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe至pe/acoet9/1)在硅胶柱上纯化粗反应产物。
得到最终产物为黄色粉末,产率67%(121mg,0.26mmol)。
1hnmr(cdcl3,400mhz):δ1.44(s,9h);1.60(s,9h);2.95(d,3h,3j=4.6hz);4.39(d,2h.3j=5.2hz);4.82(sl.1j);6.67(sl,1h);8.48(s,1h);13cnmr(cdcl3,100mhz);δ28.1(3c);28.3(4c);47.6;79.5;82.9;83.9;113.1:147.7;154.9;157.5;158.3;166.7。
实施例3.4:(6-碘-1-甲基-2,4-二氧代-2,4-二氢-1h-吡啶并[2,3-d][1,3]口恶嗪-5-基)甲基氨基甲酸叔丁酯(18)
在氮气下的25ml烧瓶中,将100mg的17(0.22mmol,1当量)溶于3ml的dcm中。同时加入342μl光气(0.65mmol,20%于甲苯中,3当量)和90μl的tea(0.65mmol,3当量),并将反应在室温下搅拌30分钟。该操作重复三次以转换所有原料。然后加入30ml水,并用dcm(3×30ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。由此获得的氮杂靛红酸酐18直接反应而无需另外的纯化步骤。
实施例4:衍生自本发明的氮杂靛红酸酐的分子与胺化合物的反应
该实施例表明,实施例1至3中合成的分子(分别为分子9、13和18)与胺化合物如苯乙胺反应。
方案6:使用苯乙胺打开本发明的氮杂靛红酸酐9、13和18。j)苯乙胺,dcm,1小时,rt。
实施例4.1:(5-碘-2-(甲基氨基)-3-(苯乙基氨基甲酰基)吡啶-4-基)甲基2-苯氧基乙酸酯(10)
向10ml烧瓶中加入65mg的9(0.13mmol,1当量)、1ml的dcm和17.4μl的苯乙胺(0.13mmol,1当量)。将反应混合物在室温下搅拌1小时。然后加入15ml水,并用et2o(5×20ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet9/1至pe/acoet7/3)在硅胶柱上纯化粗反应产物。
得到最终产物为淡黄色粉末,产率71%(50mg,0.09mmol)。
mp=164-166℃;ir(kbr)v(cm-1):3428,3296,2924,1761,1627,1433,1170,754;1hnmr(cdcl3,400mhz):δ2.86(s,3h);2.88(d,2h,3j=7.1hz);3.69(m,2h);4.57(s,2h);4.98(s,2h);5.58(d,1h,3j=4.5hz);6.54(t,1h,3j=5.6hz);6.88(d,2h,3j=8.8hz);7.00(t,1h,3j=7.6hz);7.21(m,2h);7.28(m,5h);8.40(s,1h);13cnmr(cdc13,100mhz):δ28.4;35.2;40.6;64.9;67.3;80.3;114.6(2c);118.6;122.0;126.8;128.6(2c);128.7(2c);129.7(2c);138.1;140.7;156.3;156.8;157.5;166.5;168.3。
实施例4.2:(5-碘-2-(甲基氨基)-3-(苯乙基氨基甲酰基)吡啶-4-基)甲基2-(4-氟苯氧基)乙酸酯(14)
向10ml烧瓶中加入50mg的13(0.10mmol,1当量)、1ml的dcm和12.9μl苯乙胺(0.10mmol,1当量)。将反应在室温下搅拌1小时。然后加入10ml水,并用et2o(5×15ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet7/3至pe/acoet6/4)在硅胶柱上纯化粗反应产物。得到最终产物为淡黄色粉末,产率87%(50mg,0.09mmol)。
mp=165-167℃;ir(kbr):v,3388,3376,2924,1742,1661,1577,1504,1191,824;1hnmr(cdcl3.400mhz):δ2.86(d,3h,3j=4.64hz);2.89(t,2h,3j=6.84hz);3.72(m,2h);4.51(s,2h);4.99(s,2h);5.55(d,1h,3j=4.6hz);6.51(t,1h,3j=8.0hz);6.83(m,2h);6.97(m,2h);7.21(n,3h);7.30(n,2h);8.40(s,1h);13cnmr(cdcl3,100mhz):δ28.4;35.1;40.5;65.6;67.3;80.2;115.9(d,3jc-f=8hz,2c);116.1(d,2jc-f=23hz,2c);118.6;126.8;128.6(2c);128.7(2c);138.1;140.7;153.6(d,4jc-f=2hz,1c);156.3;156.8;157.9(d,1jc-f=222hz,1c);166.5;168.1。
实施例4.3:((5-碘-2-(甲基氨基)-3-(苯乙基氨基甲酰基)吡啶-4-基)甲基)氨基甲酸叔丁酯(19)
在10ml烧瓶中,将氮杂靛红酸酐18溶于1ml的dcm中。然后加入27.6μl苯基乙胺(0.22mmol,1当量),将反应混合物在室温下搅拌1小时。然后加入10ml水,并用et2o(4×20ml)萃取溶液。然后合并有机相,用mgso4干燥并蒸发。然后使用梯度洗脱(pe/acoet7/3至pe/acoet4/6)在硅胶柱上纯化粗反应产物。
得到最终产物为淡黄色粉末,产率54%(61mg,0.12mmol)。
1hnmr(cdc13,400mhz):δ1.34(s,9h);2.79(d,3h,3j=4.6hz);2.88(t,2h,3j=7.1hz);3.67(m,2h);3.87(d,2h,3j=6.5hz);5.47(s1,1h);5.72(sl,1h);7.18(m,5h);8.27(s,1h);8.98(sl,1h);13cnmr(cdcl3,100mhz):δ28.3(3c);28.4;30.0;40.6;44.9;80.0;80.6;118.9:126.4;128.4(2c);128.7(2c);138.8,143.1;156.1;156.2;156.7;166.7。
实施例5:在热启动pcr条件下使氮杂-邻氨基苯甲酸酯化合物去保护并释放苯乙胺的方法
在此,我们证明了在实施例3中合成的衍生物10、14和19可以在热启动pcr条件下裂解并释放模拟蛋白质的苯乙胺。
一般程序:
向1.5ml管中加入:20μl的2.5mm酰胺溶液(10、14或19)的dmso溶液和180μl常规用于遗传物质扩增反应的缓冲液(60mm的trisph9,50mmko,1mm的mgcl2)。然后将混合物在95℃下在热混合器中搅拌。在t=15分钟、30分钟和1小时时进行lcms监测(条件1)。
实施例5.1:评价在95℃下氮杂-邻氨基苯甲酸苯氧基乙酸酯衍生物10的裂解
方案7:在热启动pcr条件下裂解氮杂-邻氨基苯甲酸酯化合物10
苯氧基乙酸酯基团的热去保护导致形成对应的苯甲醇(参见方案7)。然后,醇对包含在酰胺键中的羰基的亲核攻击使得能够通过产生对应的内酯而将苯乙胺释放到介质中。
该反应级联在图1的hplc色谱图中示出。
在95℃下15分钟后,将会注意到去保护的醇和预期的内酯的形成,这表明释放出了苯乙胺。该结果清楚地证明了苯氧基乙酸酯基团的热不稳定性导致形成了相应的醇。环化后,我们发现存在预期的内酯,其证明了酰胺键的裂解,并因此释放苯乙胺到介质中。在95℃下反应1小时后,初始酰胺和醇的量几乎完全消失,取而代之的是内酯形成。
该结果证明了通过该分子内环化系统在95℃裂解酰胺键的可能性。
实施例5.2:评价95℃下氟代苯氧基乙酸氮杂-邻氨基苯甲酸酯衍生物14的裂解
方案8:在热启动pcr条件下裂解氮杂-邻氨基苯甲酸盐化合物14
同样使用氟代苯氧基乙酸酯衍生物14实施去保护方法。在95℃下15分钟后,酰胺的量非常低。特别地,它低于对衍生物10观察到的结果(见图2)。氟原子的存在加速了苯氧基乙酸酯基团的裂解。因此,30分钟后,这种酰胺的量几乎完全消失,取而代之的是醇和绝大部分内酯,其表明苯乙胺被释放。
实施例5.3:评价95℃下n-boc氮杂-邻氨基苯甲酸酯衍生物19的裂解
方案9:在热启动pcr条件下裂解n-boc氮杂-邻氨基苯甲酸盐化合物19
在这种情况下,n-boc基团的去保护产生了苄胺中间体衍生物。
然后可以通过将苯乙胺释放到介质中使该衍生物环化形成对应的内酰胺(如上方案9)。显示各种中间体的hplc监测在图3中示出。
对于该衍生物,未检测到非环化的去保护形式(中间体胺)。因此,该实施例证明了去保护后n-boc基团的直接环化而形成苯乙胺。这证实,与醇官能团相比,位置5的氨基官能团具有快速环化的优点。因此,技术人员会在芳环的位置5上找到适当的用于nh2官能团保护基团,以用于获得足够快速的去保护,从而导致瞬时环化和苯乙胺释放。
实施例6:通过本发明所述的氮杂靛红酸化合物13可逆保护血红蛋白在此我们证明了:具有氟苯氧基热不稳定基团的氮杂靛红酸衍生物13能够在温和条件下在水性介质中与蛋白质反应,以形成蛋白质-氟苯氧基氮杂邻氨基苯甲酸酯衍生物。
在95℃下热处理后除去该保护基团使得能够再生天然蛋白质。
程序:
按如下所述产生混合物,其中实验al6001×至al6000.25×分别对应于在ph7.4下dmso和pbs的混合物中33μg血红蛋白与浓度增加的氮杂靛红酸化合物的反应(pbs=溶解片剂所产生的磷酸盐缓冲盐水,参考sigmap4417,在200ml水中(ph7.4))。将混合物在室温下温育2小时,然后在第120分钟利用10mm三氟乙酸的溶液中20%至72%乙腈的梯度在watersxbridgebehc4300柱(milford,usa)上收集用于hplc分析的等分试样。
表1:用于实施血红蛋白可逆酰化的试剂的相对浓度
在图4中,色谱图al602ctrl0.25×显示对照的血红蛋白未与化合物13反应。可以看到血红素和蛋白质部分的两个α和β亚基。以下三个色谱图(al6000.25×至1×)显示蛋白质部分消失,取而代之的是向右大幅偏移,其对应于由氮杂靛红化合物13酰化的α和β亚基。峰的扩展对应于蛋白质反应位点的随机酰化。
因此证明,如上所述的本发明氮杂靛红酸化合物可以与蛋白质反应。
当反应介质al6001×在pcr缓冲液(主要由trisph9组成)中在95℃下热处理15分钟时,观察到苄基酰胺部分的水解,这导致恢复天然血红蛋白的色谱图。这在图5中可见,其中上部色谱图(a)显示加入于8mguhcl中的天然血红蛋白,中间色谱图(b)显示由化合物13酰化并加入8mguhcl中的血红蛋白,下部色谱图(c)显示用化合物13酰化血红蛋白、随后在ph8的tris缓冲液中加热并加入于8mguhcl中。如前所述,可以清楚地看到血红蛋白的酰化导致形成大量酰化蛋白质,然后在热处理后该大量酰化蛋白质消失以便使天然蛋白质再生。
注意,反应介质在反应后加入于8mguhcl中,以便使在热处理过程中原本会沉淀的蛋白质部分溶解。该实施例证明了如本发明所述的化合物13对模型蛋白的可逆酰化。
实施例7:证明如本发明所述的氮杂靛红酸化合物13对taq聚合酶的可逆酰化
在此处,我们证明了:具有热不稳定的氟苯氧基的氮杂靛红酸衍生物13能够在温和条件下和在水性介质中与热稳定的聚合酶(taq)反应,以形成taq-氮杂邻氨基苯甲酸氟苯氧基衍生物。在pcr条件下在95℃热处理后除去该保护基团使得可以恢复聚合酶的活性,这通过测量其活性得到证明。
因此证明了使用适当改性的氮杂靛红酸酐临时掩盖聚合酶活性并随后在热处理后恢复其活性的概念。
程序:
我们使用了genscripttaq(2500u/100μlrefe00012),但为了消除这种酶中所有的痕量亲核试剂,我们预先通过穿过amicon10kdmicrocon管(编号42407)更换了缓冲液。因此将酶悬浮液沉积在microcon管上并离心直至缓冲液耗尽。用100μl的pbsph7.4洗涤五次,然后通过倒置管回收最后的保留物,并用pbs补足至qsp20μl以获得125u/μl的悬浮液。因此,我们证明通过该方法以非常令人满意的方式消除了tris和甘油。
然后产生如表2中所述的混合物,其中实验al6040.25×至al6040.375×分别对应于在ph7.4的dmso和pbs(pbs=由溶解片剂所产生的磷酸盐缓冲盐水,参考sigmap4417,在200ml水中(ph7.4))混合物中与浓度增加的氮杂靛红化合物反应的625单位taq聚合酶。混合物在室温下温育3小时,同时进行轻度涡旋混合,然后评估它们在热启动pcr条件下的酶活性。
表2:用于实施taq聚合酶可逆酰化的试剂的相对浓度
测定由化合物13修饰的taq聚合酶活性:
使用由“发夹”结构终止的45个碱基的寡核苷酸探针来测定taq聚合酶的聚合活性。其特征在于在结构起始位置存在荧光猝灭剂,在探针序列末端存在荧光团,使得猝灭剂在空间上接近荧光团,并且在该构型中测量不到荧光信号。由于聚合酶活性的作用,通过与前一探针的起始位置互补的19个碱基的寡核苷酸延伸该探针。由于延伸作用,探针发夹结构展开并且荧光团能够发射,然后荧光变得可测量。该荧光测量在60℃的温度下在酶活性所需的试剂存在下进行20分钟,即ph9.5的dntp、mgcl2和碱性缓冲液。
荧光的增加在测量开始时是线性的,并且能够计算对应于每分钟延伸发射的荧光量的初始速率。通过测量具有已知活性的给定聚合酶的不同浓度的u/μl的初始速率,可以建立校准曲线,使得可以测量未知活性的类似酶的活性。
通过该方法测量聚合酶的化学修饰以使其失活。通过测量酶修饰后的残留活性水平,可以确定保护的有效性。通过化学修饰完全失活的聚合酶在温度高于90℃时不会在没有热活化的情况下产生活性。
例如,通过相同的方法可容易地测量在95℃加热15分钟后恢复酶活性的能力。
因此,图6显示,取决于所选择的酰化剂浓度,taq的聚合酶活性可以完全失活并在热启动pcr条件下在95℃下处理15分钟后恢复(参见对应于al6040.375×的条件)。
- 还没有人留言评论。精彩留言会获得点赞!