用于靶向多种抗原的适配体嵌合抗原受体表达细胞的制作方法
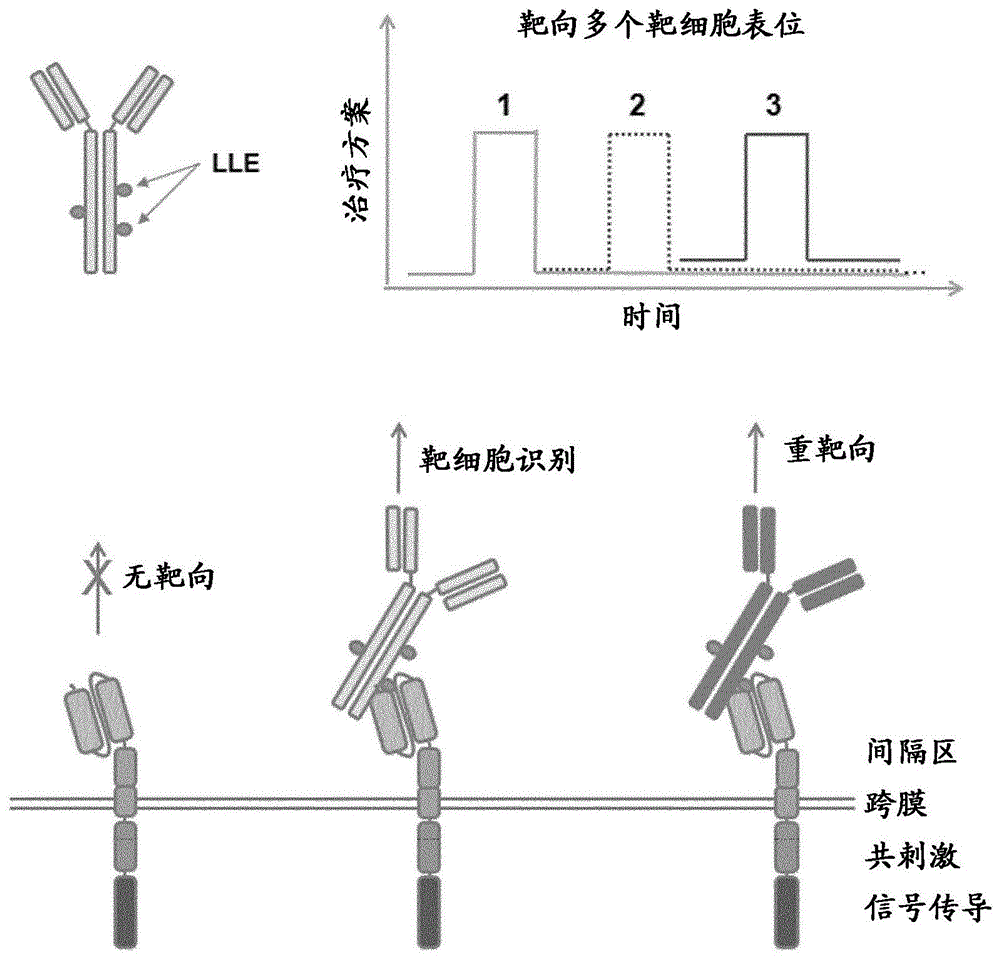
本发明一般涉及用于治疗癌症的在免疫细胞上表达的嵌合抗原受体(car)的领域,特别是适应于靶细胞的不同抗原的car。
背景技术:
通过表达转基因嵌合抗原受体将t细胞重定向至肿瘤抗原通过人白细胞抗原(hla)依赖性识别利用了有效的细胞效应机制。最近在临床试验中证实了这种方法的潜力,其中将表达car的t细胞输注到患有b细胞恶性肿瘤、神经母细胞瘤和肉瘤的成人和儿科患者中。car是通常靶向表面分子的重组受体。car通常由通过间隔区/铰链和跨膜结构域连接至可以包含共刺激结构域和t细胞激活部分的细胞内信号传导结构域的细胞外抗原识别部分组成。car独立于其主要组织相容性抗原表达识别未加工的抗原,这与生理性t细胞受体(tcr)不同。因此,cart细胞可以规避肿瘤借以躲避主要组织相容性类别(mhc)限制性t细胞识别的主要机制中的一些,例如hla表达下调或蛋白酶体抗原加工,这两种机制有助于肿瘤逃脱tcr介导的免疫力。car的另一个特征是它们不仅能够与蛋白质结合,还能与碳水化合物、神经节苷脂、蛋白多糖和高度糖基化的蛋白质结合,从而扩大了潜在靶标的范围。car通过抗原识别部分与靶标结合,所述抗原识别部分通常是衍生自抗体或fab片段的单链可变片段(scfv)(例如在daietal,2016,jnatlcancerinst108(7):djv439中综述)。
“通用”car系统允许使用相同的car来处理不同的抗原。在wo2015058018a1中,在细胞表面处表达的car结合基序以非特异性方式结合期望的靶结合分子,其中所述car的结合基序是cd16,其是非特异性地结合免疫球蛋白g抗体的低亲和性fc受体。该系统的缺点是,抗体体内充当被认为使得能够在cart细胞结合后实现特异性肿瘤靶标裂解的所施用的感兴趣的抗体的天然竞争者。
在wo2012082841a2中,公开了通用的但有适应性的抗标签嵌合抗原受体(at-car)系统,其为t细胞提供识别和杀死已被标签抗体(例如,fitc-或生物素标记的抗体)标记的靶细胞(例如肿瘤细胞)的能力和特异性。这里,缺点是标记物对于待治疗的受试者而言或者是外来分子,例如fitc,因此有免疫原性,或者标记物与内源性分子(例如生物素)相同,导致内源性分子阻断car结合基序,这阻止car结合其预期的分子标记抗体,例如生物素化抗体。
在wo2013044225a1中公开了相同的通用car系统,其面临相同的缺点。
在wo2014100615a1中,公开了一种car系统,其利用靶向未由治疗的受试者的细胞产生或表达的部分的car。因此,该car系统允许将细胞毒性淋巴细胞集中靶向靶细胞,例如癌细胞。被靶向的部分是小缀合物分子(scm)的一部分,其也包含肿瘤细胞受体的配体。因为通常使用小有机分子作为被靶向的部分,所以可以在约20分钟内实现scm从血流清除。通过施用scm以及表达car的细胞毒性淋巴细胞,淋巴细胞应答可以仅被靶向至那些表达肿瘤受体的细胞,从而减少脱靶毒性,并且淋巴细胞的激活由于scm的快速清除而可以更容易地控制。表达car的淋巴细胞还可以用作“通用”细胞毒性细胞以靶向多种多样的肿瘤而无需制备单独的car构建体。由于靶部分可以是例如fitc或生物素,该系统面临与wo2012082841a2和/或wo2013044225a1相同的缺点。
在wo2015057852a1中,公开了用于调节嵌合抗原受体效应细胞(car-ec)的活性的开关。该开关通常包含嵌合抗原受体相互作用结构域(car-id)和靶相互作用结构域(tid)。car-id可以是例如fitc或生物素。
同样,该系统面临与wo2012082841a2和/或wo2013044225a1相同的缺点。
在wo2015057834a1中,公开了一种嵌合抗原受体-效应细胞开关,其包含:a)结合效应细胞上的嵌合抗原受体的肽抗原;和b)结合靶细胞上的细胞表面分子的靶向部分。所述肽抗原是基于或衍生自天然存在的肽或非天然存在的肽。
该系统的缺点是衍生自人核蛋白的标签带来的高风险的自身免疫。
在wo2016030414a1中公开了通用嵌合抗原受体,其中所述受体包含三个结构域,其中第一结构域是标签结合结构域,第二结构域是包含细胞外铰链和跨膜结构域的连接肽链,并且第三结构域是信号转导结构域,其中标签结合结构域与衍生自任何人核蛋白的标签结合。特别地,合适的标签是来自核抗原的肽序列,其在生理条件下在天然蛋白质的上下文中不可以被相应的标签结合结构域接近和结合。该系统的缺点是标签只可以以共翻译方式加入,从而排除使用已建立的单克隆抗体作为带标签的抗原结合分子而无需事先重新设计编码所述抗原结合分子的基因。
因此,本领域需要改进的或替代的通用(适应性)car系统。
技术实现要素:
本发明公开了控制表达嵌合抗原受体(car)的细胞的功能的方法,例如表达嵌合抗原受体的免疫细胞的细胞毒性和/或免疫调节功能。如本文公开的适应性car系统包括car,所述car包含能够区分受试者中的天然存在的分子与靶细胞结合分子(tcbm)的细胞外结合结构域,所述靶细胞结合分子包含抗原结合分子(abm)、标记部分(lam)和接头部分(lim),其中所述lim缀合所述abm和所述lam(参见图1b)。所述标记部分(lam)是所述受试者中的天然存在的分子或其衍生物。所述接头部分(lim)与所述标记部分(lam)偶联。所述嵌合抗原受体以比没有接头部分的内源性标记部分更高的亲和力结合具有lam和某种lim的tcbm,由此在可能存在内源性lam的生理条件下具有提高的tcbm的识别/结合。
该方法的益处在于允许适应性car系统的lam是非免疫原性的,因为它是对于受试者为内源性的天然存在的分子。lam是自身抗原,与lim偶联的lam是修饰的自身抗原,其构建新的表位,即接头/标记表位(lle),并且lle比受试者中的天然存在的分子更好地被本文公开的car的lle结合结构域结合。使用纯自身抗原作为lam与使用修饰的自身抗原(本文公开的lle)相比具有更高的由表达car的免疫细胞触发的自身免疫的风险,所述car具有针对是自身抗原的lam的抗原结合结构域。自身免疫的风险在如本文公开的car系统中降低,因为car以比修饰的自身抗原更低的偏好(例如以较低的亲和力)结合自身抗原。
如本文公开的car系统仍然允许car免疫细胞有效募集至特异性地结合靶细胞(例如癌细胞)上表达的抗原的tcbm。天然存在的分子可以是存在于受试者的循环系统中的分子,但是以比tcbm更低的亲和力被本文公开的car结合。优先地,受试者中的天然存在的分子可以是细胞外分子或具有部分细胞外结构的分子,更优先地,受试者中的天然存在的分子可以是人非核蛋白。因此,本发明提高了治疗功效,同时使使用免疫原性lam/lim的上述风险最小化。
示例性的如本文公开的car系统显示具有是受试者中的天然存在的分子的生物素作为标记部分,具有6-(6-氨基己酰胺基)己酰基接头部分(lim),并具有以比内源性生物素更高的偏好结合6-[6-(生物素酰胺基)己酰胺基]己酰基部分的抗接头/标记表位(抗lle)car。tcbm的abm可以是可以结合靶细胞上的感兴趣的抗原的任何abm,例如肿瘤相关抗原。但并非旨在本发明受限于本实施例,因为该概念可以转移到以比并未偶联至接头部分的天然存在的分子(标记部分)更高的偏好结合与所述接头部分偶联的另外的天然存在的标记部分其他抗llecar。
本发明还公开了编码所述抗llecar的分离的多核苷酸,包含所述多核苷酸的载体,包含所述抗llecar的分离的细胞,包含所述tcbm和所述抗llecar的组合物,以及在受试者中使用所述tcbm和所述抗llecar治疗癌症的方法。
附图说明
图1:(a)接头car概念。(b)由抗原结合部分(abm)、接头部分(lim)和标记部分(lam)组成的tcbm的表示。(c)(b)的实例包含6-(6-氨基己酰胺基)己酰基接头部分(lim)和生物素标记部分(lam)和未定义的abm。
图2:测试的不同car形式的示意图(seqidno:11-20)。前导序列“l”衍生自人cd8,scfv衍生自seqidno:1和seqidno:2,铰链和间隔区“h”和“ch1”、“ch2”、“ch3”和“cd8”分别衍生自人igg4或人cd8。跨膜结构域“tm”来自人cd8,而共刺激和信号传导结构域来自4-1bb和cd3z。所有car与通过2a元件连接的标志物基因lngfr一起表达。
图3:在不同car构建体(seqidno:17、15、19、11和13)的慢病毒载体基因转移后第8天,在原代t细胞上的lngfr转基因表达。
图4:在存在或不存在10ng/ml抗cd19-生物素(4g7-生物素)抗体的情况下,未转导的(模拟)、cd19car转导的和lle-car转导的(seqidno:17、15、19、11和13)t细胞的基于萤光素酶的细胞毒性测定。在所有情况下,效应物与靶细胞的比率为1:1。显示共培养24小时后的特异性裂解,n=4。
图5:基于阻抗的实时细胞毒性测定(rtcaxcelligence)。在存在10ng/ml抗gd2-mab-生物素的情况下,对未转导的(模拟)t细胞(a)和抗llecar转导的t细胞(b)(seqidno:17、15、19、11和13)相对于ls细胞进行测试。在存在10ng/ml抗cd19-生物素抗体的情况下,对未转导的(模拟)t细胞(c)和抗llecar转导的t细胞(d)(seqidno:17、15、19、11和13)相对于mcf7-tcd19细胞进行测试。所有实验均在效应物:靶细胞比率为1:1下进行,n=3。
图6:抗llecar(seqidno:11)激活和裂解是抗体剂量依赖性的。(a)使用不同的抗gd2-mab-生物素浓度在效应物:靶细胞比率为1:1下,抗llecar相对于ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence),n=3。(b)在不同的抗cd19-mab-生物素浓度下,抗llecar(seqidno:11)相比于nalm6的基于萤光素酶的细胞毒性测定。
图7:抗llecar激活和裂解是效应物:靶细胞比率依赖性的。(a)在存在10ng/ml抗gd2-mab-生物素抗体的情况下在不同的效应物:靶细胞比率下,抗llecar(seqidno:11)相对于ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence),n=3。(b)在10ng/ml抗cd19-mab-生物素抗体下,在不同的效应物:靶细胞比率下,抗llecar(seqidno:11)相对于nalm6的基于萤光素酶的细胞毒性测定(12h),n=5。
图8:抗llecar(seqidno:11)激活和靶细胞裂解是抗原和生物素特异性的:在存在和不存在10ng/ml抗gd2-生物素(ch14.18-生物素)、抗gd2(ch14.18)、或靶细胞无关的抗-cd19-生物素(4g7-生物素)的情况下在效应物:靶细胞比率为1:1下,抗llecar相对于ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence),n=3。
图9:在存在抗cd19-生物素(4g7-生物素)、抗-cd19(4g7)的情况下,或在存在靶细胞无关的抗-gd2-生物素(ch14.18-生物素)的情况下,在效应物:靶细胞比率为1:1下,抗llecart细胞(seqidno:11)对nalm6靶细胞的特异性裂解,在24小时后通过基于萤光素酶的细胞毒性测定法测量,n=4。
图10:超生理学的生物素浓度不干扰抗llecar激活:在存在(mab)或不存在(无mab)10ng/ml抗gd2-生物素(ch14.18-生物素)抗体的情况下在不同浓度的游离生物素(如所示)下,抗llecart细胞(seqidno:11)相对于ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence)。
图11:在存在抗llecart细胞的情况下和在存在和不存在抗cd19-生物素(4g7-生物素)抗体和可变浓度的游离生物素或人ab血清的情况下,在12小时后测量的nalm6细胞的特异性裂解。
图12:(a)抗llecart细胞和通过crispr/cas9技术产生的敲除tcrαβ的抗llecart细胞的流式细胞术分析(抗tcrα/β-fitc,miltenyibiotec)。(b)通过tide确认tcrαβ敲除。(c)在存在(mab)和不存在(mab)10ng/ml抗gd2-生物素(ch14.18-生物素)抗体的情况下,抗llecart细胞(seqid11)和tcrαβ敲除的抗llecart细胞(seqidno:11)相对于ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence)。(d)在存在和不存在10ng/ml抗-tcrγδ-生物素mab的情况下在效应物:靶细胞比率为1:1下,在48小时后通过基于萤光素酶的细胞毒性测定法测量的抗llecart细胞(seqid11)和tcrαβ敲除的抗llecart细胞(seqidno:11)对molt14靶细胞的特异性裂解,n=1,4个技术重复。(e)在存在和不存在10ng/ml抗-tcrαβ-生物素mab的情况下在效应物:靶细胞比率为1:1下,在48小时后通过流式细胞术测量的与抗llecart细胞(seqidno:11)和tcrαβ敲除的抗llecart细胞(seqidno:11)一起孵育的jurkat靶细胞的相对存活,n=1,4个技术重复。
具体实施方式
本发明提供了一种用于靶向靶细胞上不同的和变化的抗原的适应性car系统,其中所述car以比受试者中的自身抗原更高的偏好结合修饰的自身抗原,从而最小化在其中使用如本文公开的car系统的受试者的自身免疫的风险。
在第一个方面,本发明提供了一种抗接头/标记表位嵌合抗原受体(抗llecar),其包含
i)细胞外接头/标记表位(lle)结合结构域,
ii)跨膜结构域,和
iii)信号转导结构域,
其中所述细胞外lle结合结构域结合靶细胞结合分子(tcbm),所述靶细胞结合分子包含
i)抗原结合部分(abm),其中所述abm特异性结合靶细胞上表达的抗原,
ii)标记部分(lam),其中所述lam是在受试者中的天然存在的分子或其衍生物,
iii)缀合所述abm和所述lam的接头部分(lim),从而形成接头/标记表位(lle),
其中所述细胞外lle结合结构域以比所述天然存在的分子更高的偏好结合所述lle。
所述抗llecar,其中所述细胞外lle结合结构域以比所述天然存在的分子高至少两倍的亲和力结合所述lle。
所述抗llecar,其中所述细胞外lle结合结构域与单体lle和所述天然存在的分子之间的结合的k(解离)值高于与多聚lle。
所述抗llecar,其中所述天然存在的分子是细胞外分子或具有部分细胞外结构的分子。
所述抗llecar,其中所述天然存在的分子是在所述受试者的循环系统中的分子。
所述抗llecar,其中所述lle是位点特异性地产生的,从而形成包含所述lam的一部分和所述lim的一部分的表位。
所述抗llecar,其中所述lam可以是所述受试者的自身抗原。
所述抗llecar,其中所述lam可以选自氨基酸、肽、蛋白质、肌酐、生物素、生物胞素、脂质、激素、维生素、碳水化合物或其衍生物。所述脂质可以是类固醇脂质,例如胆固醇。所述碳水化合物可以是例如葡萄糖、半乳糖或岩藻糖。特别优选的lam可以是生物素或其衍生物。
所述抗llecar,其中所述lim是能够产生所述lle的分子。
所述抗llecar,其中所述lim选自肽、蛋白质、核酸、碳水化合物、聚羟基烷酸酯、烷基链、烷酸、羧酸、法呢基、聚乙二醇、脂质或其衍生物。所述羧酸可以是例如是,例如ε-氨基己酸(6-氨基己酸)或6-(6-氨基己酰胺基)己酸。
特别优选的lim可以是6-(6-氨基己酰胺基)己酰基部分,例如,衍生自6-(6-氨基己酰胺基)己酸或6-(6-氨基己酰胺基)己酸活性酯,或者6-氨基己酰基部分,例如,衍生自6-氨基己酸或6-氨基己酸活性酯。
所述抗llecar,其中所述lam是生物素,并且所述lim是6-(6-氨基己酰胺基)己酰基接头部分或6-氨基己酰基接头部分。
所述抗llecar,其中所述细胞外lle结合结构域包含seqidno:1和seqidno:2的序列,由此结合6-[6-(生物素酰胺基)己酰胺基]己酰基部分。
所述抗llecar,其中被所述abm结合的所述抗原在癌细胞上表达。
所述抗llecar,其中所述抗原可以选自cd33(siglec-3)、cd123(il3ra)、cd135(flt-3)、cd44(hcam)、cd44v6、cd47、cd184(cxcr4)、clec12a(cll1)、ley、frβ、mica/b、cd305(lair-1)、cd366(tim-3)、cd96(tactile)、cd133、cd56、cd29(itgb1)、cd44(hcam)、cd47(iap)、cd66(cea)、cd112(nectin2)、cd117(c-kit)、cd133、cd146(mcam)、cd155(pvr)、cd171(licam)、cd221(igf1)、cd227(muc1)、cd243(mrd1)、cd246(alk)、cd271(lngfr)、cd19、cd20、gd2和egfr。
所述抗llecar,其中所述abm可以是抗体或其抗原结合片段。
所述抗llecar,其中所述抗llecar包含seqidno:11、seqidno:12、seqidno:13、seqidno:14、seqidno:15、或seqidno:16的序列,由此结合6-[6-(生物素酰胺基)己酰胺基]己酰基部分。优选地,所述抗llecar包含seqidno:11或seqidno:12的序列。
所述抗llecar,其中所述抗llecar在真核细胞中表达。
所述抗llecar,其中所述抗llecar在免疫细胞中表达。
所述抗llecar,其中所述抗llecar在t细胞、nk细胞、nkt细胞、γ/δt细胞或treg细胞中表达。
所述抗llecar,其中所述抗llecar在然后将分化成免疫细胞,例如将表达抗llecar的t细胞的干细胞、多谱系祖细胞或诱导多能干细胞中表达。
在另一个方面,本发明提供了编码如本文公开的抗llecar的一种或多种分离的多核苷酸。
在另一个方面,本发明提供了包含编码如本文公开的抗llecar的多核苷酸的载体或者包含编码如本文公开的抗llecar的多种多核苷酸的多种载体。
所述载体,其中所述多核苷酸可操作地连接至至少一个调节元件用于表达由所述多肽编码的所述抗llecar。
所述一种载体或多种载体,其中所述一种或多种载体是一种或多种病毒载体。
所述一种或多种载体,其中所述一种或多种病毒载体是一种或多种逆转录病毒载体或一种或多种慢病毒载体。
可以构建一种或多种核酸分子,使得它们至少编码如本文公开的car的所有实施方式。当由所述分子编码的car的所有结构域都在一个多肽内时,可以存在连续的核酸分子。或者,可以存在两个或更多个核酸分子,例如当由所述分子编码的car的一些结构域是在分开的多肽上时。
在另一个方面,本发明提供了分离的细胞,其包含如本文公开的抗llecar。
所述分离的细胞可以是真核细胞。
所述分离的细胞可以是免疫细胞,例如t细胞、nk细胞、nkt细胞、γ/δt细胞或treg细胞。
所述分离的细胞可以是干细胞、多谱系祖细胞或诱导的多能干细胞,其随后将分化成免疫细胞,例如将表达抗llecar的t细胞。
所述分离的细胞可以是t细胞,其中任选地所述t细胞不表达tcrαβ、pd1、cd3或cd96(例如,通过敲除这些基因之一)。
所述分离的细胞可以是免疫细胞,其中任选地所述细胞不表达可以是检查点、耗尽或凋亡相关的信号传导受体以及配体例如pd-1、lag-3、tigit、ctla-4、fas-l和fas-r的辅助分子(例如通过敲除这些基因中的一个)。
在另一个方面,本发明提供了分离的细胞,其包含编码如本文公开的抗llecar的一种或多种多核苷酸。
在另一个方面,本发明提供了分离的细胞,其包含含有编码如本文公开的抗llecar的多核苷酸的一种或多种载体。
在一个方面,本发明提供了组合物,其包含
a)靶细胞结合分子(tcbm),其包含
i)抗原结合部分(abm),其中所述abm特异性结合靶细胞上表达的抗原,
ii)标记部分(lam),其中所述lam是在受试者中的天然存在的分子或其衍生物,
iii)缀合所述abm和所述lam的接头部分(lim),从而形成接头/标记表位(lle),和
b)抗接头/标记表位(抗lle)嵌合抗原受体(car),所述抗llecar包含
i)细胞外接头/标记表位(lle)结合结构域,
ii)跨膜结构域,和
iii)信号转导结构域,
其中所述细胞外lle结合结构域以比所述天然存在的分子更高的偏好结合所述lle。
前文描述的tcbm和抗llecar的所有特征也可以应用于所述组合物。
在一个方面,本发明提供治疗受试者的疾病的方法,其包括
a)向需要治疗的受试者施用靶细胞结合分子(tcbm)的一种或多种制剂,所述tcbm包含
i)抗原结合部分(abm),其中所述abm特异性结合靶细胞上表达的抗原,
ii)标记部分(lam),其中所述lam是在受试者中的天然存在的分子或其衍生物,
iii)缀合所述abm和所述lam的接头部分(lim),从而形成接头/标记表位(lle),和
b)向所述受试者施用一种或多种治疗有效的抗llecar表达细胞群,所述抗llecar包含
i)细胞外接头/标记表位(lle)结合结构域,
ii)跨膜结构域,和
iii)信号转导结构域,
其中所述细胞外lle结合结构域以比所述天然存在的分子更高的偏好结合所述lle,从而诱导所述靶细胞的细胞死亡。
前文所述的tcbm和抗llecar的所有特征也可以应用于用于治疗受试者的疾病的所述方法。
所述方法,其中所述疾病的治疗是癌症,并且其中所述靶细胞是癌细胞。
所述方法,其中所述tcbm的多种制剂是相同或不同的tcbm制剂。
所述方法,其中所述多种治疗有效的抗llecar表达细胞群是相同或不同的抗llecar表达细胞群。
所述方法,其中所述多种治疗有效的群体的所述抗llecar是相同或不同的抗llecar。
所述方法,其中所述向需要治疗的受试者施用靶细胞结合分子(tcbm)的所述多种制剂是同时或随后施用。
所述方法,其中所述施用所述多种治疗有效的抗llecar表达细胞群是同时或随后施用。
本发明涉及用于治疗患有例如癌症的疾病的受试者的car系统。本发明的car系统(例如,免疫细胞,例如表达包含lle结合结构域的car的t细胞,和相应的靶细胞结合分子(tcbm))利用靶向在治疗的受试者中不出现的表位(即修饰的自身抗原)的car。由于该系统中使用的标记部分是受试者中天然存在的分子,即自身抗原,因此它是非免疫原性的。在受试者中不天然存在的表位是通过偶联标记部分与接头部分,在标记部分和接头部分的连接处产生新的界面和/或环境而产生,使得可以产生可以用作如本文公开的系统的car的接头/标记表位结合结构域的新表位识别分子。
因此,如本文公开的该car系统允许将细胞例如免疫细胞集中靶向至表达抗原的靶细胞,例如癌细胞。
所述接头部分和所述标记部分是所述靶细胞结合分子(tcbm)的一部分,所述靶细胞结合分子也包含抗原结合部分(abm),其中所述接头部分缀合lam和abm。通常,所述abm针对靶细胞表面上表达的抗原。
通过施用tcbm以及表达car的免疫细胞,免疫细胞应答可以仅靶向在靶细胞表面上表达抗原的那些细胞,从而降低脱靶毒性。表达car的免疫细胞可用作“通用”免疫细胞以靶向多种靶细胞,例如多种肿瘤,而无需制备单独的car构建体。由car识别的tcbm的标记/接头表位也可以保持恒定。只需要改变tcbm的abm以允许系统靶向不同特性的靶细胞。
以下说明如本文公开的car系统的示例:
生物素是受试者(例如人)的循环系统中的天然存在的分子。因此,生物素对于所述受试者是非免疫原性的。
制备tcbm,其包含生物素作为标记部分与6-(6-氨基己酰胺基)己酰基接头部分偶联(缀合),产生6-[6-(生物素酰胺基)己酰胺基]己酰基部分。
tcbm还包含abm,例如对靶细胞表面上的抗原(例如在癌细胞上表达的抗原gd-2或cd19)特异的抗体或scfv,其中6-(6-氨基己酰胺基)己酰基接头部分桥接生物素和abm。转导免疫细胞以表达包含6-[6-(生物素酰胺基)己酰胺基]己酰基部分结合结构域作为lle结合结构域的car。因此,该car靶向上述tcbm。
将两种组分,即tcbm和表达car的免疫细胞,施用于患有例如癌症的疾病的受试者。tcbm由靶细胞结合,例如癌细胞(通过tcbm的abm部分与关联靶细胞抗原例如gd-2或cd19的结合)。然后tcbm的6-[6-(生物素酰胺基)己酰胺基]己酰基部分被免疫细胞表达的抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分car识别并结合。在6-[6-(生物素酰氨基)己酰胺基]己酰基-abm缀合物结合后,表达抗-6-[6-(生物素酰胺基)己酰胺基]己酰基部分car的免疫细胞被激活,靶细胞,例如癌细胞,可以被杀死。抗6-[6-(生物素酰氨基)己酰胺基]己酰基部分car以比受试者中的循环生物素更高的偏好结合6-[6-(生物素酰胺基)己酰氨基]己酰基缀合物。
如本文公开的表达car的免疫细胞不可以通过直接结合靶细胞如癌细胞来杀死细胞。因此,tcbm充当表达car的免疫细胞和靶细胞之间的桥梁。只要产生tcbm的接头/标记表位的标记/接头部分是未天然见于受试者中的部分并且car的细胞外lle结合结构域以比所述天然存在的分子更高的偏好结合所述lle,则表达car的免疫细胞的活性可以被限制于靶细胞。此外,可以通过限制施用于受试者的tcbm的量来调节表达car的免疫细胞的激活,例如,如果检测到副作用,则通过操纵tcbm的输注。
本发明的car系统利用tcbm作为表达car的免疫细胞和表达抗原的靶细胞之间的桥梁。tcbm包含通过接头部分连接的在分子的一端的标记部分(lam)和在另一端的抗原结合部分(abm)。对标记部分的特性的唯一要求仅在于其必须是受试者中天然存在的分子(自身抗原)并且其缀合的接头部分的变体可以被免疫细胞表达的car识别和结合,所述car以比非缀合的接头部分(天然存在的变体)更高的亲和力与其缀合的接头部分的变体(修饰的自身抗原)结合。abm可包含单一抗原特异性(单特异性)、两种或更多种抗原特异性(双特异性和多特异性)。双特异性abm的实例是与抗原cd19和cd20结合的abm。
abm还可以包含单价结合以及二价和多价结合位点。二价abm的实例是包含识别抗原的不同表位的两个scfv的abm。
abm可以是针对衍生自宿主(例如小鼠)的免疫的靶标的免疫球蛋白。此外,它还可以衍生自合理设计和重组表达。在另一种实施方式中,abm还可以衍生自例如定向进化和筛选技术例如噬菌体展示和核糖体展示的方法。标记部分和抗原结合部分可以通过接头部分共价缀合,例如生物素和6-(6-氨基己酰氨基)己酰基部分的化学偶联。
或者,标记部分和抗原结合部分可以通过接头部分非共价缀合。此外,或者,tcbm可以在abm和lim之间包含其他部分或结构域。唯一的要求是lim以使得允许产生如本文公开的接头/标记表位的方式与lam缀合(或偶联)。
接头部分可以与标记部分缀合,产生包含lle的部分。与lam偶联的lim可以例如通过使用例如lim的氨基反应性琥珀酰亚胺酯衍生物靶向蛋白abm上的赖氨酸而随机偶联。
或者,接头部分可以与abm位点特异性地缀合,例如通过使用例如在所述abm的编码序列中的工程化终止密码子处递送非天然氨基酸的无义抑制子trna,从而在重组表达的蛋白abm中引入所述非天然氨基酸。
可能能够产生如本文公开的lle的每个分子可以用作接头部分。对接头部分的特性的唯一要求是接头部分可以被化学缀合(或偶联)到标记部分或被遗传(重组)编码,并且应该能够在如本文公开的接头部分和标记部分的上下文、界面和/或环境处产生新表位。lim可以优先是在受试者中不引起或不倾向于引起免疫反应的分子,例如lim是自身抗原。在这种情况下,lam和lim的界面产生新的表位,即lle。
lim可以例如选自肽、蛋白质、核酸、碳水化合物、聚羟基烷酸酯、烷基链,烷酸、羧酸(例如,ε-氨基己酸(6-氨基己酸)或6-(6-氨基己酰胺基)己酸)、法呢基、聚乙二醇、脂质及其衍生物。
特别优选的lim可以是6-(6-氨基己酰胺基)己酰基部分,例如衍生自6-(6-氨基己酰胺基)己酸或6-(6-氨基己酰胺基)己酸活性酯,或6-氨基己酰基部分,例如衍生自6-氨基己酸或6-氨基己酸活性酯。
所述抗llecar,其中所述lam是生物素,并且所述lim是6-(6-氨基己酰胺基)己酰基接头部分或6-氨基己酰基接头部分。
本领域技术人员将理解并认识到,可以使用各种手段来制备由标记部分、连接部分和抗原结合部分组成的tcbm。
在施用至受试者之前,tcbm可以制备成药学上可接受的制剂。这样的制剂可含有药学上可接受的赋形剂或载体。
abm结合靶细胞上的抗原的亲和力可以变化,但通常结合亲和力可以是至少约100μm、1nm、10nm或100nm,优选至少约1pm或10pm,甚至更优选至少约100pm。
要求如本文公开的car的细胞外lle结合结构域以比相应天然存在的分子更高的偏好结合tcbm的lle。
所述car的细胞外lle结合结构域可以使用可商购抗体或其抗原结合片段制备。
或者,可以通过本领域技术人员公知的方法新生产用于选择的lle的抗体或抗原识别分子。
car的lle结合结构域结合tcbm的lle的亲和力可以变化,但通常结合亲和力可以在0.01nm至1μm的范围内,优选在0.1-100nm的范围内,或更优选地在1-30nm的范围内。唯一的要求是在受试者中car的lle结合结构域结合tcbm的lle的亲和力必须高于car的lle结合结构域结合天然存在的分子(即没有接头部分)的亲和力。优选地,所述细胞外lle结合结构域以比所述天然存在的分子高至少两倍,优选至少5倍,更优选至少10倍,最优选至少高20倍的亲和力结合所述lle。
编码本发明的car的一种或多种dna或rna构建体(一种或多种核酸分子)可通过本领域公知的方法(例如,包括逆转录病毒和慢病毒的基于病毒的系统、包括电穿孔的物理方法、生物学方法、化学方法)转染或转导到宿主细胞中。不管用什么方法在宿主细胞中整合,优选稳定整合,编码本发明的car的dna,结果是宿主细胞表达如本文公开的car。
或者,核酸序列可以合成产生。
在转染/转导过程后可以分离(富集或分离)如本文公开的表达car的工程化细胞,用于通过本领域公知的方法(例如基于荧光的分离技术,例如
通常,可以从受试者获得细胞,例如免疫细胞,优选t细胞,用于产生本发明的表达car的工程化细胞。细胞,例如免疫细胞,优选t细胞,可以从多种来源获得,例如外周血单核细胞(pmbc)、骨髓、淋巴结组织、脐带血或胸腺组织。为了富集这些细胞,可以使用本领域公知的方法,例如通过ficolltm或percolltm梯度离心或阳性/阴性选择技术,例如荧光分选(例如fcassort)或磁性分选(例如
示例性地,受试者的血液样品的t细胞被磁性地标记,例如用磁珠偶联至对cd4特异的抗体,洗涤,磁性富集和收集。然后可以将这些t细胞工程化以在其细胞表面上表达如本文公开的car。
在本发明的一种实施方式中,表达如本文公开的car的分离/富集的工程化细胞,例如免疫细胞,优选t细胞,可以在遗传修饰之前或之后激活,并且使用本领域公知的方法扩增以增加工程化(t)细胞的数量,例如用由与人源化cd3和cd28激动剂缀合的胶体纳米基质组成的transactt细胞试剂(miltenyibiotec;wo2014048920a1)对t细胞进行多克隆刺激。优选地,所述工程化细胞(例如免疫细胞,例如t细胞)的数量可以增加至治疗有效量。
如本文公开的的表达car的细胞可以通过编码本发明的car的rna进行工程化。可以通过本领域公知的方法(例如基于病毒的系统、物理方法、生物学方法、化学方法)将rna转染或转导到宿主细胞中。无论用什么方法将编码本发明的car的rna转移到宿主细胞中,结果是宿主细胞表达如本文公开的car。“rna工程化细胞”在有限时间内表达car(瞬时表达)。
表达如本文公开的car的遗传修饰(免疫)细胞,优选t细胞,可以在封闭系统的自动化过程中产生。一种用于产生遗传修饰细胞,优选t细胞的方法,所述方法可以可包括例如以下步骤:
a)提供包含(免疫)细胞的细胞样品,
b)通过离心准备细胞样品,
c)磁性分离(免疫)细胞,优选t细胞,
d)使用调节剂激活富集的(免疫)细胞,优选t细胞,
e)遗传修饰(免疫)细胞,优选t细胞,以表达本发明的car,
f)在培养室中扩增遗传修饰的(免疫)细胞,优选t细胞,
g)洗涤培养的(免疫)细胞,优选t细胞。
所有这些步骤可以在封闭系统中自动执行,优选地在封闭和无菌系统中执行。
该方法特别适用于制备基因修饰的细胞,例如免疫细胞,优选t细胞,其中富集的(免疫)细胞,优选t细胞,通过使用病毒和/或非病毒载体进行基因修饰。
这些步骤中的任何一个都可以增加、省略或者可以以不同的顺序发生。
调节剂可选自激动性抗体和/或细胞因子。
基因修饰的(免疫)细胞,优选t细胞,可以通过(免疫)细胞的磁性标记和培养之前或之后的磁性分离来富集以在最终的细胞产品中获得更高频率的基因修饰(免疫)细胞,优选t细胞。
作为用于细胞修饰的封闭且无菌的系统,可以使用全自动细胞处理装置clinimacs
如本文公开的car系统可用于治疗患有与抗原相关的疾病、病症或状况的受试者,所述抗原可以被如本文公开的tcbm的抗原结合部分特异性地结合。
特别地,如本文公开的car系统可用于治疗患有癌症的受试者。可以分离受试者的细胞,例如免疫细胞,例如t细胞,或者可以使用建立的免疫细胞系。受试者可以患有所述癌症(患者)或可以是健康的受试者。这些免疫细胞在体外被遗传修饰以表达如本文公开的car。这些工程化细胞可以在体外激活并扩增至治疗有效的抗llecar表达细胞群。在细胞疗法中,除了第二药物组合物,tcbm制剂外,这些工程化细胞可以作为药物组合物(或治疗有效的抗llecar表达细胞群的制剂)输注给有需要的接受者。所述接受者中的输注的细胞可以能够杀死(或至少停止生长)表达被如本文公开的car系统识别的抗原的癌细胞。接受者可以是从其获得细胞的同一受试者(自体细胞疗法)或可以是相同物种的另一个受试者(同种异体细胞疗法)。
可以在向受试者施用tcbm制剂之前将治疗有效的抗llecar表达细胞群施用于患者。或者,可以在向受试者施用治疗有效的抗llecar表达细胞群之前或同时将tcbm制剂施用于受试者。另外的变化包括在施用至受试者之前,体外培养治疗有效的抗llecar表达细胞群与tcbm制剂的tbcm。
可以使用本领域技术人员已知的技术配制抗lle-car表达(免疫)细胞群以施用至受试者。
包含治疗有效的抗llecar表达细胞群的制剂可包含一种或多种药学上可接受的赋形剂(载体或稀释剂)。包含在制剂中的赋形剂将具有不同的目的,这取决于例如抗lle-car的lle结合结构域的性质,所使用的免疫细胞的(亚)群体,和施用方式。通常使用的赋形剂的实例包括但不限于:盐水、缓冲盐水、右旋糖、注射用水、甘油、乙醇及其组合、稳定剂、增溶剂和表面活性剂、缓冲剂和防腐剂、张度剂、填充剂和润滑剂。
抗llecar表达细胞的一个或多个治疗有效群体的制剂可包含抗lle-car表达(免疫)细胞的一个群体,或抗lle-car表达(免疫)细胞的多于一个群体。抗lle-car(免疫)细胞的不同群体可以基于lle结合结构域的特性、激活结构域的特性、免疫细胞的(亚)群体的特性,或其组合而变化。
可以使用本领域技术人员已知的方式和技术将包含抗llecar表达细胞的一个或多个治疗有效群体的制剂施用于受试者。示例性方式包括但不限于静脉内注射。其他方式包括但不限于肿瘤内、皮内、皮下(s.c,s.q、sub-q、hypo)、肌肉内(i.m.)、腹膜内(i.p.)、动脉内、髓内、心内、关节内(关节)、滑膜内(关节液区域)、颅内、脊柱内和鞘内(脊髓液)。
施用至受试者的包含抗llecar表达细胞的一个或多个治疗有效群体的制剂包含有效治疗特定适应症或疾病的数量的抗lle-car表达细胞,例如免疫细胞。通常,可以施用包含约1×104至约1×1010个抗lle-car表达细胞(例如免疫细胞)的制剂。在大多数情况下,所述制剂可以包含约1×105至约1×109个抗lle-car表达细胞,例如免疫细胞,约5×105至约5×108个抗lle-car表达细胞,例如免疫细胞,或约1×106至约1×107个抗lle-car表达细胞,例如免疫细胞。然而,施用于受试者的抗lle-car表达细胞(例如免疫细胞)的数量可在宽范围内变化,这取决于疾病的位置、来源、特性、程度和严重程度、待治疗的个体的年龄和状况等。医生可以最终确定要使用的合适剂量。
可以使用本领域技术人员已知的技术配制tcbm用于施用于受试者。tcbm的制剂可包含一种或多种药学上可接受的赋形剂(载体或稀释剂)。包含在所述制剂中的赋形剂具有不同的目的,这取决于例如标记部分、接头部分、abm的性质和施用方式。通常使用的赋形剂的实例包括但不限于:盐水、缓冲盐水、右旋糖、注射用水、甘油、乙醇及其组合、稳定剂、增溶剂和表面活性剂、缓冲剂和防腐剂、张度剂、填充剂和润滑剂。
tcbm的制剂可包含一种类型的tcbm,或多种类型的tcbm。不同类型的tbcm可基于标记部分、接头部分、抗原结合部分或其组合的特性而变化。
可以使用本领域技术人员已知的方式和技术将tcbm施用于受试者。示例性方式包括但不限于静脉内、腹膜内和肿瘤内注射。其他方式包括但不限于皮内、皮下(s.c,s.q、sub-q、hypo)、动脉内、髓内、心内、关节内(关节)、滑膜内(关节液区域)、颅内、脊柱内和鞘内(脊髓液)。
将包含tcbm的制剂以有效治疗特定适应症或疾病的量施用至受试者。通常,可以将包含至少约1μg/kg至约100mg/kg体重的tcbm的制剂施用给需要治疗的受试者。在大多数情况下,考虑到施用途径、症状等,剂量可以是每天约100μg/kg至约10mg/kg体重的tcbm。如图6所示,令人意外的是,使用如本文公开的car系统,可以使用浓度低至1ng/ml(对应于1μg/kg体重)的tcbm。然而,施用至受试者的制剂中的tcbm的量可在宽范围内变化,这取决于疾病的位置、来源、特性、程度和严重程度、待治疗的个体的年龄和状况等。医生可以最终确定要使用的合适剂量。
car表达细胞制剂和tcbm制剂的施用之间的时间选择可以为宽范围,这取决于多种因素,包括所使用的(免疫)细胞类型、car的结合特异性、标记部分、接头部分和抗原结合部分的特性、靶细胞(例如待治疗的癌细胞)的特性、靶细胞在受试者中的位置、用于向受试者施用制剂的手段,以及接受治疗的受试者的健康、年龄和体重。实际上,tcbm制剂可以在遗传工程化(免疫)细胞制剂之前、同时或之后施用。
取决于治疗的疾病,施用car表达细胞制剂的步骤,或施用tcbm制剂的步骤,或两者,可重复一次或多次。当两种或更多种tcbm制剂可以应用于受试者时,所应用的制剂可以包含相同或不同的tcbm。当两种或更多种工程化细胞制剂例如本发明的表达car免疫细胞被应用于受试者时,工程化细胞可以是相同细胞类型或不同细胞类型。细胞制剂例如免疫细胞也可包含一种以上的细胞类型,各自表达本发明的car。
定义
除非另外定义,否则本文使用的技术和科学术语具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。通常,car可包含包含抗原结合结构域的细胞外结构域(细胞外部分)、跨膜结构域和细胞内信号传导结构域。细胞外结构域可以通过接头与跨膜结构域连接。细胞外结构域还可包含信号肽。本发明的car的细胞外部分包含如本文公开的接头/标记表位(lle)结合结构域。具体地,如本文公开的car具有细胞外lle结合结构域作为抗原结合结构域。
“信号肽”是指指导蛋白质在细胞内转运和定位的肽序列,例如转运和定位至某种细胞器(例如内质网)和/或细胞表面。
通常,“抗原结合结构域”是指特异性结合抗原(从而能够靶向含有抗原的细胞)的car的区域。本发明的car可包含一个或多个抗原结合结构域。通常,car上的靶向区域是在细胞外的。抗原结合结构域可包含抗体或其抗原结合片段。抗原结合结构域可包括,例如,全长重链、fab片段、单链fv(scfv)片段、二价单链抗体或双抗体。任何与给定抗原特异性结合的分子(例如来自天然存在的受体的亲和体或配体结合结构域)可用作抗原结合结构域。抗原结合结构域通常是scfv。通常,在scfv中,免疫球蛋白重链和轻链的可变区通过柔性接头融合以形成scfv。这样的接头可以是例如“(g4/s1)3-接头”。
在某些情况下,抗原结合结构域衍生自其中将使用car的相同物种是有益的。例如,当计划在人体内治疗性地使用它时,car抗原结合结构域包含人或人源化抗体或其抗原结合片段是有益的。人或人源化抗体或其抗原结合片段可通过本领域公知的多种方法制备。如本文公开的car具有细胞外接头/标记表位结合结构域作为抗原结合结构域,允许通过如本文公开的靶细胞结合分子间接地结合靶细胞上表达的抗原。
如本文所用,“间隔区”或“铰链”是指位于抗原结合结构域和跨膜结构域之间的亲水区。本发明的car可以包含细胞外间隔区结构域,但也可以去掉这种间隔区。间隔区可包括例如抗体的fc片段或其片段、抗体的铰链区或其片段、抗体的ch2或ch3区、辅助蛋白、人工间隔区序列或其组合。间隔区的突出实例是cd8α铰链。
car的跨膜结构域可以衍生自这样的结构域的任何期望的天然或合成来源。当来源是天然的时,结构域可以衍生自任何膜结合或跨膜蛋白。跨膜结构域可以衍生自例如cd8α或cd28。当关键信号传导和抗原识别模块(结构域)在两个(或甚至更多个)多肽上时,car可具有两个(或更多个)跨膜结构域。由于car的每个多肽中的小分子依赖性异二聚化结构域,分裂的关键信号传导和抗原识别模块使得能够对car细胞表达进行小分子依赖性、可滴定和可逆的控制(wuetal,2015,science350:293-303)。
car的细胞质结构域(或细胞内信号传导结构域)负责激活其中表达car的免疫细胞的至少一种正常效应功能。“效应功能”是指细胞的特定功能,例如在t细胞中,效应功能可以是细胞溶解活性或辅助活性,包括细胞因子的分泌。细胞内信号传导结构域是指转导效应物功能信号并指导表达car的细胞执行专门功能的蛋白质的部分。细胞内信号传导结构域可包括足以转导启动或阻断免疫细胞效应功能的信号的给定蛋白质的细胞内信号传导结构域的任何完整的、突变的或截短的部分。
用于car的细胞内信号传导结构域的突出实例包括t细胞受体(tcr)的细胞质信号传导序列和在抗原受体结合后启动信号转导的共受体。
通常,t细胞激活可以由两种不同类型的细胞质信号传导序列介导,首先是通过tcr启动抗原依赖性初级激活的那些(初级细胞质信号传导序列),其次是以抗原非依赖性方式起作用以提供次级或共刺激信号的那些(次级细胞质信号传导序列,共刺激信号传导结构域)。因此,car的细胞内信号传导结构域可包含一个或多个初级细胞质信号传导结构域和/或一个或多个次级细胞质信号传导结构域。
以刺激方式起作用的初级细胞质信号转导序列可含有itam(基于免疫受体酪氨酸的激活基序信号传导基序)。
通常用于car的包含初级细胞质信号序列的itam的实例是衍生自tcrζ(cd3ζ)、fcrγ、fcrβ、cd3γ、cd3δ、cd3ε、cd5、cd22、cd79a、cd79b和cd66d。最突出的是衍生自cd3ζ的序列。
car的细胞质结构域可以设计为自身包含cd3-ζ信号传导结构域或与一个或多个任何其他期望的细胞质结构域组合。car的细胞质结构域可包含cd3ζ链部分和共刺激信号传导区域。共刺激信号传导区域是指包含共刺激分子的细胞内结构域的car的一部分。共刺激分子是除了淋巴细胞对抗原的有效响应所需的抗原受体或其配体之外的细胞表面分子。共刺激分子的实例是cd27、cd28、4-1bb(cd137)、ox40、cd30、cd40、pd-1、icos、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3。
car的细胞质信号传导部分内的细胞质信号传导序列可以以随机或指定的顺序彼此连接,有或没有接头。长度优选为2-10个氨基酸的短的寡肽或多肽接头可形成连接。突出的接头是甘氨酸-丝氨酸双联体。
例如,细胞质结构域可包含cd3-ζ的信号传导结构域和cd28的信号传导结构域。在另一个实例中,细胞质结构域可包含cd3-ζ的信号传导结构域和cd27的信号传导结构域。在另一个实例中,细胞质结构域可包含cd3-ζ的信号传导结构域、cd28的信号传导结构域和cd27的信号传导结构域。
如上所述,出于car的分裂的关键信号传导和抗原识别模块的目的,car的细胞外部分或跨膜结构域或细胞质结构域也可包含异二聚化结构域。
本发明的car,即包含lle结合结构域的car,可以被设计为以任何顺序和/或组合包含如本文所述的上述结构域的任何部分或部分,产生功能性car。
本文使用的术语“抗体”以最广泛的含义使用以涵盖各种形式的抗体结构,包括但不限于特异性识别(即结合)靶抗原的单克隆和多克隆抗体(包括全长抗体)、多特异性抗体(例如双特异性抗体)、抗体片段(即抗体、免疫粘附素和抗体-免疫粘附素嵌合体的抗原结合片段)。“抗体片段”包含全长抗体的一部分,优选其可变结构域,或至少其抗原结合位点(“抗体的抗原结合片段”)。抗体片段的实例包括fab(片段抗原结合)、scfv(单链片段可变)、单结构域抗体、双抗体、dsfv、fab’、双抗体、单链抗体分子和由抗体片段形成的多特异性抗体。
如本文所用,术语“抗原”旨在包括结合或引起一种或多种抗体产生的物质,并且可包括但不限于蛋白质、肽、多肽、寡肽、脂质、碳水化合物和其组合,例如,糖基化蛋白质或糖脂。如本文所用的术语“抗原”是指可以在靶细胞上表达并且可以被包括但不限于抗体或tcr、或工程化分子(包括但不限于转基因tcr、car、scfv或其多聚体、fab片段或其多聚体、抗体或其多聚体、单链抗体或其多聚体、或可以以高亲和力执行结合至结构的任何其他分子)的适应性免疫系统识别的分子实体。
本文所用的术语“抗-6-[6-(生物素酰胺基)己酰胺基]己酰基部分car”是指结合tcbm的car,所述tcbm含有生物素作为lam,含有衍生自例如6-(6-氨基己酰胺基)己酸或6-(6-氨基己酰胺基)己酸活性酯的6-(6-氨基己酰胺基)己酰基接头部分作为lim,并且含有针对靶细胞的抗原的abm。
6-(6-氨基己酰胺基)己酰基接头部分和生物素标记部分的界面、连接和/或相互作用部分产生接头/标记表位(lle),即6-[6-(生物素酰氨基)己酰胺基]己酰基部分。
例如,将生物素-lc-lc-nhs(thermofisherscientific,mw567,70g/mol,目录号21343,cas-no.89889-52-1)偶联至如本文公开的abm导致可以被如本文公开的所述抗-6-[6-(生物素酰氨基)己酰胺基]己酰基部分car结合的如本文公开的tcbm。
如例如图1c中所示的6-(6-氨基己酰胺基)己酰基接头部分可以具有任何功能性化学基团,例如羧基、活性酯、乙酰胺或马来酰亚胺,其能够使用用于偶联至其的nh2或sh基团偶联至如本文公开的abm(例如抗体或其片段)。
如本文所用的术语“靶细胞”是指在其细胞表面上表达抗原的细胞,其应被如本文公开的靶细胞结合分子的抗原结合部分识别(结合)。靶细胞可以是癌细胞或任何其他疾病细胞。
术语“疾病”是指受试者中的功能异常或紊乱,例如癌症、自身免疫疾病或病毒、细菌、寄生虫或其他的感染。
如本文公开的car(多肽)、编码car的核酸分子、重组表达载体、表达car的细胞和表达car的细胞群,可以被分离和/或纯化。术语“分离的”意指从天然状态改变或移除。例如,分离的细胞群意味着这些细胞的富集和从通常在天然存在状态下与所述分离的细胞相关的其他细胞中分离。分离的细胞群是指基本上纯化的细胞群,其是比在自然界中发现的更均质的细胞群。优选地,富集的细胞群包含至少约90%的所选择的细胞类型。在特定方面,细胞群包含至少约90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或甚至100%的所选的细胞类型。
例如,天然存在于活动物中的核酸或肽不是“分离的”,但是从其天然状态的共存材料部分或完全分离的相同核酸或肽是“分离的”。分离的核酸或蛋白质也可以存在于非天然环境中,例如存在于宿主细胞中。
如本文所用,术语“受试者”是指哺乳动物,例如小鼠、大鼠、牛、猪、山羊、鸡、猴或人。优选地,受试者是人。受试者可以是患有例如癌症的疾病的受试者(患者),但受试者也可以是健康受试者。
如本文所用的术语“自体的”是指衍生自相同受试者的任何材料,其随后被重新导入。
如本文所用的术语“同种异体的”是指衍生自相同物种的与重新导入材料的受试者不同的受试者的任何材料,
术语“治疗有效量”或“治疗有效群体”是指在受试者中提供治疗益处的细胞群的量。
与抗体、如例如在本文公开的tcbm中使用的其抗原结合片段、或car的抗原结合结构域相关的术语“特异性结合”或“特异于”是指识别并结合特定抗原但基本上不识别或结合样品中的其他分子的抗原结合结构域。特异性结合来自一个物种的抗原的抗原结合结构域也可以结合来自另一个物种的抗原。这种跨物种反应性对于许多抗体是典型的,因此不与该抗原结合结构域是特异性的的定义相矛盾。特异性结合抗原的抗原结合结构域也可以结合抗原的不同等位基因形式(等位基因变体、剪接变体、同种型等)或来自相同基因家族的该抗原的同源变体。这种交叉反应性对于许多抗体是典型的,因此不与该抗原结合结构域是特异的的定义相矛盾。
如本文所用的术语“工程化细胞”和“遗传修饰的细胞”可互换使用。该术语意指含有和/或表达进而改变细胞或其后代的基因型或表型的外源基因或核酸序列。特别地,该术语是指细胞,优选免疫细胞,可以通过本领域公知的重组方法操作以稳定或瞬时表达在天然状态下不在这些细胞中表达的肽或蛋白质。例如,免疫细胞被工程化以在其细胞表面上表达人工构建体,例如嵌合抗原受体。例如,可以使用逆转录病毒或慢病毒载体将car序列递送到细胞中。
术语“免疫细胞”或“免疫效应细胞”是指可以是免疫系统的一部分并且执行特定效应功能的细胞,例如α-βt细胞、nk细胞、nkt细胞、b细胞、先天性淋巴细胞(ilc)、细胞因子诱导的杀伤(cik)细胞、淋巴因子激活的杀伤(lak)细胞、γ-δt细胞、间充质干细胞或间充质基质细胞(msc)、单核细胞或巨噬细胞。优选的免疫细胞是具有细胞毒性效应功能的细胞,例如α-βt细胞、nk细胞、nkt细胞、ilc、cik细胞、lak细胞或γ-δt细胞。“效应功能”是指细胞的特定功能,例如在t细胞中,效应功能可以是细胞溶解活性或辅助活性,包括细胞因子的分泌。
本文所用的术语“治疗”(治疗)疾病是指降低受试者经历的疾病或病症的至少一种体征或症状的频率或严重性。
如本文所用的术语“表达”定义为由其启动子在细胞中驱动的特定核苷酸序列的转录和/或翻译。
术语“表位”是指可被免疫系统,具体地被抗体、b细胞或t细胞识别的抗原部分。例如,表位是抗体或其抗原结合片段结合至其的抗原的特定片段。
如本文所用的术语“接头/标记表位”(lle)或“标记/接头表位”可互换使用,并且是指由本文公开的tcbm的缀合的接头部分和标记部分的上下文、界面和/或环境形成的表位。通过标记部分与接头部分的偶联产生的表位在受试者中不是天然存在的。产生的表位包含所述lam的一部分和所述lim的一部分。优选地,所述lle在旨在用表达如本文公开的car的细胞治疗的受试者中不引起或不倾向于引起免疫反应。对lle的唯一要求是它是与包含lle结合结构域的car相关的表位。如本文公开的衍生自表位识别分子(例如识别标记/接头表位的抗体)的car的细胞外lle结合结构域以比没有接头部分的内源性标记部分(即受试者中的天然存在的分子)(自身抗原)更高的偏好结合新产生的表位(即标记/接头表位)(修饰的自身抗原)。
所述car的所述细胞外lle结合结构域以比所述天然存在的分子高至少两倍,优选至少5倍,更优选至少10倍,最优选至少高20倍的亲和力结合所述lle。
如序列表方案中给出的seqidno:1至seqidno:20的氨基酸序列主要是如本文公开的car的部分或完整序列。所述seqidno:1至seqidno:20的序列还可以分别包含这些序列的变体,其具有缺失、增加或替换的一些氨基酸,同时仍保留如本文所述的预期功能。因此,该定义中包括的是分别为seqidno:1至seqidno:20的氨基酸序列的变体,例如分别基本上类似于seqidno:1至seqidno:20的氨基酸序列,在氨基酸序列水平上具有至少70%、或至少75%、80%、85%、90%、95%、97%、98%或99%的序列同一性的分别。通常,在该定义中包括分别不导致序列seqidno:1至seqidno:20的预期功能的有意改变的所有氨基酸变异。在本发明的上下文中,“序列同一性”可以使用本领域公知用于氨基酸序列的比对程序使用成对比对来确定。
“循环系统”是受试者的器官系统,其允许血液将营养物(例如氨基酸和电解质)、氧气、二氧化碳、激素和血细胞循环并输送往返体内细胞以提供营养并帮助对抗疾病,稳定温度和ph值,并维持体内平衡。循环系统包括两个独立的系统:分配血液的心血管系统和循环淋巴的淋巴系统。
如本文所用,术语“受试者中的天然存在的分子或其衍生物”是指受试者中的分子或物质,优选地,所述分子位于细胞外或至少具有细胞外部分,例如跨膜蛋白。天然存在的分子可以以游离形式存在或者共价或非共价结合另一分子,例如结合蛋白质。例如,生物素以游离的形式在血液系统中循环,但也结合例如血浆蛋白。
由于这种要求,这些分子是非免疫原性的,因为它们是受试者的内源性分子(自身抗原)。例如,生物素是受试者中天然存在的分子,因为它是受试者的血液系统(循环系统)中的循环分子。术语“其衍生物”在本文中意指受试者的所述天然存在的分子可以经历一些微小的修饰而不改变所述分子的性质。所述修饰与所述分子的接头部分的缀合不相同。
实施方式
除了如本文公开的car系统的上述应用之外,在下文中描述本发明的其他实施方式,但不旨在限于这些实施方式。
在本发明的一种实施方式中,抗llecar是抗6-[6-(生物素酰氨基)己酰胺基]己酰基部分car,其中所述细胞外lle结合结构域包含seqidno:1和seqidno:2的序列,tcbm包含6-[6-(生物素酰胺基)己酰胺基]己酰基部分和特异性地结合癌细胞抗原(例如抗gd2或cd19)的abm。
本文提供的结果显示,无论选择的abm如何,抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分car的car系统和包含6-[6-(生物素酰氨基)己酰氨基]己酰基部分的tcbm很好地起作用。
已经产生了多种版本的抗-6-[6-(生物素酰胺基)己酰胺基]己酰基部分car结构,具有包含seqidno:1和seqidno:2的scfv的可变重-轻链方向(hl和lh),以及可变细胞外间隔区序列(图2),并且已经将这些序列克隆到能够编码car分子以及标志物(lngfr)的多顺反子慢病毒载体骨架中。
图3显示,对于所有测试的构建体,病毒载体的产生和lngfr的表达都是可能的。
从pbmc富集t细胞,并使用编码如图2所示的构建体的慢病毒载体在体外激活和转导。然后将不同的抗-6-[6-(生物素酰胺基)己酰胺基]己酰基部分cart细胞与ls在1:1的比率下共培养。将缀合6-[6-(生物素酰胺基)己酰胺基]己酰基部分的抗cd19抗体以给定剂量加入培养物中或不加入(阴性对照)。然后测量由抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分cart细胞引起的细胞毒性。证实seqidno:11是最有效的构建体(图4)。
使用不同的肿瘤细胞系和不同的抗体(分别为mcf7和抗cd19)确认结果,如图5a-d所示。
抗gd2或抗cd19抗体的剂量滴定显示抗-6-[6-(生物素酰胺基)己酰胺基]己酰基部分cart细胞在低至1ng/ml的剂量下有效杀死其肿瘤靶标(图6a和b)。该剂量远低于体内抗肿瘤功能所需的剂量。细胞毒性进一步随不同的效应细胞与靶细胞的比率而变化(图7a和b)。
在不存在标记(例如生物素化)抗体或存在对肿瘤特异的非标记抗体或存在对肿瘤不特异的标记抗体的情况下,抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分cart细胞可以对肿瘤靶标不具有细胞毒性。这是使用2种不同模型的如图8和图9所示的情况。抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分car功能必须不受血液中存在的潜在交叉反应性配体例如循环生物素的损害。人血清中存在的生物素的剂量范围为60至370pmol/l(mocketal,1995,jofnutrition)。显示在存在高达30μg/ml的生物素的剂量(远超生物素的生理水平)或高达50%的人ab血清的情况下,抗6-[6-(生物素酰胺基)己酰胺基]己酰基部分car的功能不受损害。参见图10和图11。
在本发明的另一种实施方式中,如本文公开的抗llecar在来自供体(同种异体)或患者(自体)的t细胞中表达。激活这些细胞并进行基因修饰(使用病毒载体或非病毒载体)以表达抗llecar。将这些基因修饰的t细胞作为治疗有效的抗llecar表达细胞群输注到患者体内。向患者输注确定剂量的tcbm制剂,所述tcbm制剂包含针对表达被tcbm识别的抗原的靶细胞的abm。
tcbm可以在输注抗llecart细胞输注之前、期间和/或之后递送至接受者。
然后,抗llecart细胞将识别与其特定靶标结合的tcbm。进而,抗llecart细胞对结合的tcbm的识别诱导输注的修饰的t细胞提供其预期的功能-主要是表达细胞因子和/或是细胞毒性的(图1)。
例如,tcbm可以靶向恶性细胞表面的抗原,使得抗llecart细胞能够杀死肿瘤细胞。在另一个应用中,tcbm输注的由感染特定病原体的细胞表达的靶抗原,并使t细胞能够介导杀死感染的细胞或由基因修饰的t细胞释放炎性细胞因子。在另一个例子中,tcbm结合细胞表面抗原或在炎症部位表达的分子,以诱导来自t细胞的调节响应。t细胞响应的差异可以通过使用的t细胞的类型(例如细胞毒性cd8+t细胞或调节性cd4+t细胞)和用于产生抗llecar的细胞内信号传导结构域的类型来控制(例如cd3ζ和4-1bb以诱导促炎响应或cd3ζ和pd-1以诱导抗炎响应)。
为了提供增强的控制和安全性,可以以剂量、空间和时间方式使用tcbm的制剂之一或组合。
可以进行tcbm的剂量响应,由此患者接受抗llecart细胞和极低剂量的tcbm,然后连续增加tcbm的剂量直至观察到期望的响应或直至达到剂量限制性毒性。
可以输注多种tcbm以靶向在一种或多种靶细胞上表达的多于一种靶抗原,有效地产生多克隆响应。
具有不同abm的tcbm可以一个接一个地递送。
在脱离tcbm递送后,抗llecart细胞不再能够提供其期望的效应功能。但是,在重新输注tcbm后可以恢复该功能。
如本文公开的car系统还可以以下列方式使用:第一波tcbm输注到患者的靶免疫调节细胞(例如treg、m2、tam、mdsc、骨髓衍生细胞、单核细胞)中。并且连续波的tcbm靶向肿瘤以被破坏。这种方法的目的是首先破坏免疫屏障,防止t细胞仅在时间限制性的时间窗口内有效地攻击肿瘤。
在本发明的另一种实施方式中,抗llecar在非常有效但在体内寿命短的nk细胞和/或nkt细胞中表达。这种选择细胞作为基因修饰的效应细胞是旨在用于其中体内基因修饰细胞的存在仅在规定的时间段(例如少于6个月,或甚至至少于2个月)内需要的治疗。
如本文公开的抗llecar也可以在γ/δt细胞中表达,以获益于其效应物和归巢能力。该情况尤其适用于从供体到受体的同种异体转移以避免gvhd的情况。
在本发明的一种实施方式中,如本文公开的结合抗llecar的tcbm可以包含abm中的两个不同的抗原识别位点(本文称为“双特异性”,而不是对于相同抗原为二价),其中tcbm的每个识别位点具有低亲和力和/或高koff,因此靶标必须表达抗原两者,以使“二价”tcbm以足够的亲合力结合。仅表达一种靶标的细胞将不允许“二价”tcbm足够好地结合抗llecart细胞以递送其效应功能。
在另一种方法中,tcbm靶向肽mhc复合物。
如本文公开的抗llecar还可以在干细胞和/或多谱系祖细胞和/或诱导多能干细胞中表达,优选由它们自身的启动子驱动,优选仅在t细胞中有活性的谱系特异性启动子。然后细胞将从基因修饰的干细胞和/或多谱系祖细胞分化,并产生将表达抗llecar及其自身t细胞受体的t细胞。该策略优选用于患者进行同种异体干细胞移植的情况。一旦干细胞和/或多谱系祖细胞产生t细胞,就可以通过输注特定tcbm来驱动针对靶向的疾病的免疫响应。
或者,使用能够特异性靶向适应性免疫系统的亚集例如cd8和/或cd4t细胞、nk、nkt细胞和/或γδt细胞的修饰病毒载体,在体内直接修饰患者的t细胞。
如本文公开的抗llecar还可以在总t细胞、cd8阳性t细胞或cd4阳性t细胞中表达,并与如本文公开的标记/接头部分(例如6-[6-(生物素酰胺基)己酰胺基]己酰基部分)缀合的抗体和/或fab的文库组合使用以发现应该使用靶向基序(例如抗体)的哪种组合来杀死给定的靶细胞。然后可以使用适当的组合来治疗给定患者的疾病(即,指导的组合car疗法)。
在本发明的另一种实施方式中,产生表达如本文公开的抗llecar并且不表达tcrαβ的t细胞以产生“通用-通用”(u2)cart细胞。例如,可以以这种方式对将分化成t细胞的干细胞进行基因工程化,在患者中产生修饰的t细胞的完整组。修饰的t细胞可以以同种异体移植的形式施用给患者。u2抗llecar方法符合通用靶向的标准,因为抗llecart细胞激活的特异性完全由tcbm的预定义的特异性决定。此外,tcrαβ链或cd3的敲除与t细胞受体复合物的连续表达失败,在不存在gvhd保证的情况下促进了在供体通用性方面的通用靶向,并且进一步地,其促进了tcrαβ+和cd3+阳性细胞的靶向,而没有通过car靶向进行的自毁。
在本发明的另一种实施方式中,免疫细胞表达如本文公开的抗llecar,并且另外抑制或已经敲除了可以是检查点、耗尽或凋亡相关的信号传导受体以及配体例如pd-1、lag-3、tigit、ctla-4、fas-l和fas-r的辅助分子,而且抗llecar细胞中cd4或cd8的敲除将允许瞬时靶向cd4+或cd8+细胞,这可以是治疗自身免疫疾病例如移植物抗宿主病、实体器官排斥、原发性自身免疫疾病:t细胞和b细胞相关的自身免疫(类风湿性关节炎、慢性炎症性肠病、多发性硬化症、重症肌无力等)的有吸引力的选项。
如上所述,如本文公开的抗llecar可以在细胞例如免疫细胞中表达以靶向癌细胞的抗原以治疗癌症。可以治疗的癌症包括未血管化或尚未显著血管化的肿瘤,以及血管化肿瘤。癌症可包括非实体肿瘤(例如血液肿瘤,例如白血病和淋巴瘤)或可包括实体瘤。用如本文公开的car系统治疗的癌症的类型包括但不限于癌、胚细胞瘤和肉瘤、以及某些白血病或淋巴样恶性肿瘤、良性和恶性肿瘤,和恶性肿瘤(例如肉瘤、癌和黑素瘤)。还包括成人肿瘤/癌症和儿科肿瘤/癌症。
血液癌的实例包括白血病,包括急性白血病(例如急性淋巴细胞白血病、急性髓细胞白血病、急性髓性白血病和成髓细胞、早幼粒细胞、髓单核细胞、单核细胞和红白血病)、慢性白血病(例如慢性粒细胞(粒细胞)白血病、慢性髓性白血病和慢性淋巴细胞白血病)、真性红细胞增多症、淋巴瘤、霍奇金病、非霍奇金淋巴瘤(惰性和高级形式)、多发性骨髓瘤、瓦尔登斯特伦巨球蛋白血症、重链疾病、骨髓发育不良综合征、毛细胞白血病和骨髓发育不良。
实体瘤是通常不包含囊肿或液体区域的异常组织块。实体瘤可以是良性或恶性的。不同类型的实体瘤以形成它们的细胞类型命名(例如肉瘤、癌和淋巴瘤)。实体瘤的实例,例如肉瘤和癌,包括纤维肉瘤、粘液肉瘤、脂肪肉瘤、软骨肉瘤、骨肉瘤和其他肉瘤、滑膜瘤、间皮瘤、尤因瘤、平滑肌肉瘤、横纹肌肉瘤、结肠癌、淋巴恶性肿瘤、胰腺癌、乳腺癌、肺癌、卵巢癌、前列腺癌,肝细胞癌、鳞状细胞癌、基底细胞癌、腺癌、汗腺癌、甲状腺髓样癌、甲状腺乳头状癌、嗜铬细胞瘤、皮脂腺癌、乳头状癌、腺乳头状癌、髓样癌、支气管源癌、肾细胞癌、肝癌、胆管癌、绒毛膜癌、wilms肿瘤、宫颈癌、睾丸肿瘤、精原细胞瘤、膀胱癌、黑色素瘤和中枢神经系统肿瘤(例如胶质瘤(例如脑干胶质瘤和混合性胶质瘤)、胶质母细胞瘤(又称多形胶质母细胞瘤)、星形细胞瘤、中枢神经系统淋巴瘤、生殖细胞瘤、成神经管细胞瘤、神经鞘瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、少突胶质瘤、网膜瘤、神经母细胞瘤、视网膜母细胞瘤和脑转移)。
如本文公开的抗llecar可以在免疫细胞中表达以靶向与病毒、细菌、寄生虫或其他感染相关的抗原以治疗感染。
如本文公开的抗llecar可以在细胞例如免疫细胞中表达以靶向自身抗原以治疗自身免疫疾病。
然而,如本文公开的car系统不应解释为仅限于如本文公开的抗原靶标和疾病。相反,它应被解释为包括与其中如本文公开的car系统可用于治疗疾病的疾病相关的任何抗原靶标。
实施例
实施例1.抗llecart细胞的产生
1.1编码抗llecar的不同间隔区变体的慢病毒转运载体构建体的文库的产生
为了产生识别生物素的抗llecar以及将生物素与识别靶细胞表位的抗体连接的接头部分的部分,基于抗biomb抗体的可变重链(vh)和可变轻链(vl)(seqidno:1和2)在vh-vl和vl-vh方向上设计scfv(单链可变片段)。可变链通过柔性(g4s)3接头(seqidno:4)连接,人cd8前导序列(seqidno:3)置于scfv的n末端。优化序列以在人宿主细胞中表达,并通过dna2.0合成,bamhi和nhei切割位点分别在scfv序列的5’和3’侧翼。将这些scfv亚克隆到含有基于人igg4(hc)和人cd8茎的不同间隔区变体、2a元件以及表达标志物(lngfr)的转移载体质粒中(图2)。在大肠杆菌neb5α中扩增质粒dna,并根据制造商的方案使用qiagenendofreeplasmidmaxikit制备。通过sanger测序(gatc)验证所有序列。
1.2lv颗粒的产生
为了产生慢病毒颗粒,使用25ml培养基(dmem、10%(v/v)fcs、2mml-谷氨酰胺)将0.5×105/cm2hek293t细胞接种在t175细胞培养瓶(175cm2)中并于37℃,5%co2下孵育过夜。在第二天贴壁细胞通常表现出75-90%汇合度。将培养基更换为含有2mml-谷氨酰胺、不含fcs的dmem,并制备转染dna混合物(使用lipofectamine3000,thermofisher试剂方案)。为此,使用0.2μg/cm2(对于一个t175培养瓶为35μg)的总dna量,并制备两种单独的混合物a(dna)和b(lipofectamine)。对于混合物a:转移载体质粒0.073μg/cm2(12.7μg)、辅助质粒1(vsvg)0.018μg/cm2(3.18μg)、辅助质粒2(gag/rev/pol)0.109μg/cm2(19.09μg)、p3000增强剂试剂2μl/μg总dna,和opti-mem0.033ml/cm2通过短暂涡旋混合。对于混合物b:将lipofectamine30000.75/cm2重悬于opti-mem0.033ml/cm2中。将混合物a和b分别孵育5分钟,然后将混合物a和b轻轻混合在一起,并在室温下孵育20分钟。然后将脂质/dna复合物逐滴加到接种的细胞中并在37℃下孵育约12小时。此后,用0.133ml/cm2新鲜培养基(dmem、10%fcs、2mml-谷氨酰胺)替换培养基,并在37℃、5%co2下孵育24小时。通过移液小心地除去细胞培养物上清液来收获组装的慢病毒颗粒。通过以5000×g离心10分钟从上清液中除去细胞碎片。然后将含lv的上清液直接用于转导t细胞,在4℃下储存过夜或在液氮中快速冷冻并储存在-80℃。对于第二轮慢病毒颗粒生产,向转染的hek293t细胞中加入0.133ml/cm2新鲜培养基(dmem、10%fcs、2mml-谷氨酰胺),并在37℃,5%co2下再继续孵育24小时。24小时后,如前所述收获上清液。
1.3抗llecart细胞的转导、扩增和表达分析
从健康供体抽取血液样品,并通过在biocoll(biochrom)上分离而产生pbmc。使用macs技术(miltenyibiotec)选择cd4+和cd8+t细胞。对于t细胞激活和扩增,根据制造商的说明使用t细胞激活/扩增试剂盒,人(miltenyibiotec)。将t细胞接种到24孔板中,每孔2mlt细胞悬浮液,浓度为在含有人ab血清(10%(v/v)gemcell)、il-7(10ng/ml)和il-15(5ng/ml)的texmacs培养基中1×106细胞/ml。
在激活后第2天进行转导。为此,将lv上清液以3的moi加入t细胞中,并通过上下移液小心地重悬浮细胞。在激活后第8天,根据制造商的说明,使用在ls柱(miltenyibiotec)上的抗lngfr微珠通过macs分离表达lngfr的细胞。使用t细胞激活/扩增试剂盒,人(miltenyibiotec)重新激活选择的细胞,并以1×106个细胞/ml(每孔2ml)的浓度接种在24孔板中。然后将细胞连续扩增,并在其第一次激活后第14天通过facs分析测定转导效率。为此,取出等分试样,并根据制造商的说明使用抗lngfrapc(miltenyibiotec)对lngfr表达进行染色(图3)。
实施例2.tcbm产生
根据本领域公知的方案通过在氨基处的琥珀酰亚胺基酯进行随机标记而进行抗体修饰。对于修饰,通过流经在缓冲液中平衡的sephadexg25柱,将抗体重新缓冲至peb缓冲液中。使用bradford测定法测定收集的级分的蛋白质含量。合并含蛋白质的级分并测定总体积。通过280nm处的吸收测量最终蛋白质浓度。随后,将相应量的生物素-lc-lc-nhs(thermofisherscientific,分子量567,70g/mol,目录号21343,cas号89889-52-1)溶解在dmso中。将抗体和dmso/标记物混合物在室温(21℃)孵育1小时,然后流经sephadexg25柱。再次收集级分,测定蛋白质浓度并合并含蛋白质的级分。通过280nm处的吸收测量最终蛋白质浓度。通过孵育表达抗体靶标的抗体细胞系和用荧光染料缀合的抗生物素抗体进行二次染色,然后进行facs分析,确认成功的生物素化。此外,haba测定用于测定每个抗体的生物素分子数量。
实施例3.用于细胞毒性分析的靶细胞系的产生
3.1稳定转导的萤光素酶表达细胞系的产生
根据1.2,lv颗粒的产生是类似的。肿瘤细胞转导方案与t细胞(1.3中描述)类似。使用的转移载体质粒pcdh-ef-effly-t2a-mcherry和pcdh-ef-effly-t2a-gfp由helmholtzzentrummunchen(德国环境卫生研究中心,基因载体部门,binjevick博士和irmelajeremiaspd医学博士)慷慨提供。转导后,将肿瘤细胞扩增(upscale)并通过流式细胞术评估转导效率(gfp激发488nm激光,带通滤波器525/50nm;mcherry激发561nm激光,带通滤波器615/20nm)。通过facs细胞分选中-高(gfp/mcherry)群体而富集细胞,丢弃阴性、低阳性和高阳性细胞亚组。将中-高(gfp/mcherry)肿瘤细胞系再次扩增,评估(gfp/mcherry)荧光,冷冻保存并用于功能分析。用pcdh-ef-effly-t2a-mcherry和/或pcdh-ef-effly-t2a-gfp稳定转导以下细胞系:均为b细胞前体白血病细胞系的nalm-6和nalm-16,均为t细胞急性淋巴细胞白血病细胞系的tcrγδ阳性molt14和tcrαβ阳性jurkat,以及均为神经母细胞瘤细胞系的ls和lan-1。
3.2表达截短的cd19的mcf-7转染子的产生
通过转染线性化的、截短的cd19dna构建体,通过在冰上在imdm培养基(iscove'smodifieddulbecco'smedium,thermofisherscientific)中电穿孔,稳定地转染贴壁乳腺癌细胞系mcf-7(michigancancerfoundation-7)细胞系。电穿孔在200v电压和975μf下进行,时间常数在40至80之间。对于选择,抗生素g418(geneticin,invivogen)以1mg/ml使用。通过基于facs的细胞分选,分离具有中-高tcd19表达的均匀表达的mcf-7,并进行扩增以进行冷冻保存和功能分析。
实施例4.基于xcelligence实时细胞分析(rtca)装置的杀死测定4.1用于xcelligence的ls培养
-培养3天(最佳)
-5866个细胞/cm2->中间细胞培养瓶(75cm2)调节至在15ml培养基中为0.44×106个细胞或大细胞培养瓶(175cm2)调节至在35ml培养基中为1×106/ml。
-仅使用60%和80%汇合度的细胞
收集和接种:
a)丢弃培养基(rpmi1640、10%fcs、1%青霉素/链霉素、5%l-谷氨酰胺)
b)每个t175cm2烧瓶加入3ml胰蛋白酶/edta并孵育2分钟
c)加入8ml完全培养基,小心分配细胞并以300g离心5分钟
d)将细胞置于10ml新鲜培养基中并用台盼蓝溶液计数细胞
e)根据xcelligence方案接种细胞
4.2xcelligence杀死测定方案
第0天:
-expnotes:命名实验并将其保存在指定管理员(数据目录)下
-布局:命名孔
-时间表:添加步骤1(一次)用于校准(自动调整),添加步骤2(间隔30分钟(15分钟),完成测量时间48h(100h)
rtca设备的校准:
-填充培养基50μl/孔,将板放入测量设备并按下开始按钮以启动校准。完成测量后取出板(“准备下一步”)
靶标:
-将ls(之前培养三天)调整为0.15×106/ml
-在100μl中以15000个细胞/孔接种肿瘤细胞,
-加入100μl培养基/孔
-将板放回测量设备并按下开始按钮
-记录开始时间
第1天:
24小时后,细胞指数在1-3之间。按下停止按钮,将板从测量设备中取出以继续进行实验。如果细胞指数低于1,则丢弃实验。
30分钟内移液:
a)小心取出100μl培养基(剩余体积为100μl)
b)以50μl体积添加效应细胞
c)以50μl体积添加mab
d)所有孔中的总体积:200μl(在无mab条件下补满培养基)将板置于测量装置中并继续数据采集。
强制性对照:
-背景:每4孔仅培养基,+/-mab
-每4孔仅靶标,+/-mab
实施例5.基于萤光素酶的杀死测定
5.1背景
d-萤光素是萤火虫萤光素酶的双发光反应的底物。它与生存力线性相关,与靶细胞死亡负相关。
5.2d-萤光素-等分试样的制备
a)将10mgd-萤光素(合成的,sigmaaldrich,目录号l950410mg)溶解在1mldmso中,以达到10mg/ml的浓度
b)加入24mldmso→浓度为0.4mg/ml
c)准备55μl等分试样(10%额外体积)
d)储存在-20℃
5.2.1使用
a)从55μl等分试样中取出50μl,并加入在实验中使用的5ml培养基(cave:用于人血浆实验,仅使用rpmi,无fcs)→1:100稀释→浓度为0.004μg/μl=4μg/ml
b)如果移液体积为50μl并且每个孔中的最终体积为200μl
→1:4稀释,终浓度为1μg/ml
5.3材料、细胞、试剂和仪器
a)稳定转导的萤火虫萤光素酶阳性靶细胞系
b)效应细胞
c)rpmi培养基,merck-millipore,目录号:f1215-500ml
d)平底白板,greinerbioone,目录号:655083
e)d-萤光素合成,目录号:l9504,sigmaaldrich
f)advia120siemensbayer(细胞计数)
g)perkinelmerwallac1420victor2微板读数器(读板器)
5.4程序
a)靶细胞制备:(对一块板的计算)
用advia计数靶细胞用于测定。从细胞储备液中取10e06个细胞,以300g离心10分钟,完全除去上清液,调整细胞数为0.1×10e6个细胞/50μl。
为了保证每孔低孔偏差,第1列和第12列用作100%对照,仅肿瘤细胞。此外,生物发光滴定100%、75%、50%、25%、10%和0%的靶细胞用于在excel中通过线性趋势线计算来计算裂解公式。
b)效应细胞制备
用advia计数效应细胞,并加入50μl培养基中。
c)在50μl培养基中以预定浓度在相应条件下加入抗体。
d)根据“d-萤光素等分试样的制备”添加d-萤光素。
e)将板置于37℃,5%co2的培养箱中。
f)在6、12、24和48小时测量生物发光。
g)仪器设置:生物发光的采集,37℃,victorwallac读数器。
实施例6.细胞内细胞因子染色(ics)
6.1缓冲液/试剂
-pbs-edta:500mlpbs、2mmedta(当你有5m的浓度时为2ml)
-facs缓冲液:500mlpbs、2%fcs(10ml)、2mmedta(当你有5m的浓度时为2ml)、0.02%naazid(当你有10%的浓度时为1ml)
-permwash:500mlpbs、0.1%皂苷-s7900(0.5g)、0.05%bsaa-3059(2.5g)、0.02%naazid(当你有10%的浓度时为1ml)
-布雷菲德菌素(brefeldin)asigma(1:125)和/或莫能菌素(monensin),例如golgistop
-布雷菲德菌素建议用于细胞内细胞因子染色,莫能菌素用于cd107a
-活/死着色:invitrogen
-cytofix/cytoperm:bd
-感兴趣的抗体
-96孔圆底板
6.2程序
-每隔一个孔使用孔以避免洗涤程序中的污染
-总孵育体积200μl
-孵育时间16小时
开始实验:在每个孔中加入50μl布雷菲德菌素a。
需要补偿:未染色的细胞(与实验中使用的完全相同)。它们在测定期间以相同的方式处理,但不染色。
染色1-3
开始前30分钟准备:
-将离心机冷却至4℃
-准备缓冲液
-获取冰以将板存放在冰上
-配制活/死染色溶液:50μl/孔,溶液1:400
孵育时间之后
-离心板(1800rpm,2min,4℃)
-用200μlpbse洗涤(1800rpm,2min,4℃)
-用200μlpbse洗涤(1800rpm,2min,4℃)
a)加入50μl/孔的活/死溶液并重悬细胞(不是未染色的状态)
在4℃(黑暗)下孵育时间20分钟
在此期间:产生细胞外主混合物(mastermix)(在facs缓冲液中),全部在一个falcon中
在l/d溶液的孵育时间后:
-加入150μlfacs缓冲液(1800rpm,2min,4℃)
-用200μlfacs缓冲液洗涤(1800rpm,2min,4℃)
b)加入50μl/孔细胞外主混合物并重悬细胞(既不是未染色的状态,也不是同型的状态)
在4℃(黑暗)下孵育时间20分钟
孵育时间后:
-加入150μlfacs缓冲液(1800rpm,2min,4℃)
-用200μlfacs缓冲液洗涤(1800rpm,2min,4℃)
-加入100μl/孔cytofix/cytoperm并重悬其
在4℃(黑暗)下孵育时间20分钟
在此期间:产生细胞内主混合物(在permwash中),全部在一个falcon中
孵育时间后(cytofix/cytoperm):
-加入100μl/孔permwash(1800rpm,2min,4℃)
-用200μlpermwash洗涤(1800rpm,2min,4℃)
c)加入50μl/孔细胞内主混合物并重悬其(不是未染色的)
在4℃(黑暗)下孵育时间20分钟
在此期间:产生pfa溶液(总是新的,不保留)、facs缓冲液+1%多聚甲醛、储备液浓度37%,200μl/孔
孵育时间(iz)后:
-加入150μlpermwash(1800rpm,4℃,2分钟)
-用200μlpermwash洗涤(1800rpm,2min,4℃)
-用200μlpermwash洗涤(1800rpm,2min,4℃)
-加入200μl/孔pfa并重悬其
储存在4-8℃直至facs分析
实施例7.使用crispr/cas9技术产生抗llecar+tcrαβ-t细胞
根据上述方案,从抗llecart细胞(seqidno:11)产生抗llecar+tcrαβ-细胞。使用
7.1使用
材料/技术:
a)如1.3中所示产生抗llecart细胞(seqidno:11)。
b)cart细胞培养基(texmacs,10%人血清,10ng/mlil7,5ng/mlil15)
c)dpbs-dulbecco磷酸盐缓冲盐水
d)定制的修饰的grna(用于tcr敲低的指导rna序列已经在文献中描述并且应该是本领域技术人员可获得的)
e)cas9mrna从tebu-bio订购,目录号:l-6125-100
f)
-电解缓冲液e
-重悬缓冲液t
-10μl
-
程序:
a)在微量离心管中转移2e06cart细胞(seqidno:11)
b)在微型离心机中以800g,5分钟,用1mlpbs缓冲液洗涤cart细胞(seqidno:11)细胞3次,并小心去除上清液
列出使用的试剂:
a)用缓冲液e(3ml)填充电穿孔管
b)加载程序
c)使用100μl电穿孔吸头,并且每次电穿孔取100μl的相应样品
d)使用100μl缓冲液t校准电穿孔装置neon
e)如果自检批准,没有错误,则继续样品电穿孔
电穿孔设置:电压为1.400,脉冲为10,脉冲数为3,吸头类型为100μl
f)对于每次电穿孔,将电穿孔移液管放入电穿孔管中。
g)开始电穿孔过程
h)电穿孔后,立即取出
通过流式细胞术和t7测试(实施例8)进行成功敲除的检测。
实施例8.抗llecar修饰的t细胞的功能分析
从pbmc富集t细胞,并使用编码所示构建体的慢病毒载体在体外激活和转导。然后将不同的抗llecart细胞与肿瘤细胞(nalm6)以1比1的比率共培养。将生物素化的抗cd19抗体以给定剂量加入培养物中或不加入(阴性对照)。然后测量由抗llecart细胞和cd19cart细胞引起的细胞毒性(图4)。表达由seqidno:11编码的car的cart细胞显示出最大的细胞毒性作用,与直接抗cd19cart细胞(阳性对照)相当。结果表示从四个不同供体一式三份进行的四次独立实验的平均值。使用实时细胞毒性测定(rtcaxcelligence)和不同的肿瘤细胞系确认结果。抗llecart细胞在生物素化的抗gd2抗体存在下对ls细胞显示出细胞毒性(图5b),再次由seqidno:11编码的car给出最有效的细胞毒性响应。此外,抗llecart细胞在生物素化的抗cd19抗体存在下表现出针对mcf-tcd19细胞的细胞毒性(图5d)。针对不同肿瘤相关抗原(例如cd19和gd2)的mab滴定显示出在100pg/ml-10μg/ml靶标特异性标记的mab浓度范围内的有利的抗llecart活性(图6a和b)。该剂量明显低于体内有效治疗应用所需要的抗体剂量。在注册于clinicaltrials.gov的临床试验“ch14.181021antibodyandil2afterhaplosctinchildrenwithrelapsedneuroblastoma”(nct02258815)中,抗gd2mabch14.18在连续5天以20mg/m2施用,导致高于20μg/ml的高mab血清浓度(未发表的数据,研究招募中)。因此,在我们的患者个体同情使用中,在高风险b系前体all患者中使用fc优化的嵌合抗cd19mab治疗(4g7sdie),观察到高mab浓度。以20mg/m2每2周应用4g7sdie。实际上,在体内输注抗cd19抗体后,据报道血液中可检测到60-90μg/ml的水平(molther.2016,pmid:27380762)。针对靶向滴定的效应物显示出线性相关性(图7a和b)。
此外,使用两种不同的模型,抗-llecart细胞在不存在标记抗体的情况下,或在存在对肿瘤特异的非标记抗体的情况下,或在存在对肿瘤不特异的标记抗体的情况下,对肿瘤靶细胞显示出低细胞毒性(图8、图9)。xcelligencertca如4中概述用神经母细胞瘤细胞系ls(表达gd2的肿瘤细胞系)以效应物比靶标的比率为1:1进行。在该实验中,在仅有肿瘤细胞系而没有效应细胞(抗llecart细胞),有抗llecart细胞而没有mab的情况下,以及在抗llecart细胞与未标记的gd2特异性mabch14.18和抗llecart与结合cd19(其不被ls表达)的特异性标记mab(4g7sdie)的情况下,仅观察到轻微差异(图8)。然而,仅在抗llecart加上标记的gd2特异性mabch14.18的情况下在不到24小时内观察到ls的完全裂解。24小时后,不可以从抗llecart细胞排除一些轻微的非特异性抗肿瘤活性(图8)。在基于萤光素酶的测定中获得,仅在存在抗cd19-生物素(4g7-生物素)的情况下,通过抗llecart细胞优先裂解nalm6靶细胞的类似结果(图9)。
最后,抗llecar功能必须不受血液中存在的潜在交叉反应性配体例如循环生物素损害。显示在存在高达30μg/ml的生物素的剂量(远超生物素的生理水平)或高达50%的人ab血清的情况下,抗llecar的功能不受损害(图10和11)。xcelligencertca如4中概述用神经母细胞瘤细胞系ls(表达gd2的肿瘤细胞系)和抗llecart细胞以效应物比靶标的比率为1:1在不存在或存在游离生物素(其在细胞培养基中以低(生理)浓度加到超生理浓度)的情况下进行。无论游离生物素浓度如何,在使用抗llecart细胞和10ng/mlgd2特异性mabch14.18的测试条件下,在不到24小时内观察到肿瘤细胞的完全根除(图10)。此外,在用b细胞前体白血病细胞系nalm-6进行的细胞毒性测量期间未观察到抗llecart细胞的抑制(图11)。生理和超生理浓度的游离生物素或人ab血清都不降低抗llecart细胞介导的裂解的特异性裂解(图11)。
为了产生“通用-通用”(u2)cart细胞,如实施例7中所述产生抗lle-cart(seqidno:11)tcrαβ敲除细胞。通过tcrαβ表达分析确认成功的敲除(图12a和b)。根据常规染色方案进行流式细胞术。将细胞在含有人白蛋白的pbs缓冲液中洗涤两次,以300g离心10分钟(加速/制动9/9),在黑暗中于4℃染色15分钟,然后再次洗涤两次,然后在根据制造商的方案用抗小鼠补偿珠(bdbiosciences,订购号552843)进行补偿后根据制造商的方案在bd-lsrii上采集。使用tcrabfitc(miltenyibiotec,抗tcrα/β-fitc,抗人(克隆:bw242/412),订购号130-098-690),lngfrapc(miltenyibiotec,cd271(lngfr)-apc,抗人(克隆:me20.4-1.h4),订购号130-091-884),cd8apc-cy7(biolegend,抗人(克隆:ski),订购号344713),cd4uv1buv395(bdbiosciences,(克隆:rpa-t4)订购号564724)。虽然cd4/cd8比率保持不变,但tcrαβ表达的降低清楚可见(图12a)。该结果通过t7和tide确认,其根据本领域公知的方案进行(图12b)。在存在和不存在10ng/ml抗gd2-生物素(chl4.18-生物素)抗体的情况下进行抗llecart细胞(seqidno:11)和tcrαβ敲除的抗llecart细胞(seqidno:11)相对ls的基于阻抗的实时细胞毒性测定(rtcaxcelligence),其显示通过crispr/cas9技术产生的tcrαβ敲除的抗llecar(seqidno:11)t细胞在体外细胞溶解中表现得与tcrαβ阳性抗llecar(seqidno:11)t细胞一样好(图12c)。此外,在通过抗llecart细胞(seqidno:11)和tcrαβ敲除的抗llecart细胞(seqidno:11)对molt14靶细胞进行特异性裂解的过程中,通过表达或不表达tcrαβ链的抗llecart细胞观察到类似的靶标裂解(图12d)。此外,当在存在10ng/ml抗tcrαβ-生物素mab的情况下孵育48小时时,在效应物:靶细胞比率为1:1下,tcrαβ敲除的抗llecart细胞显著降低jurkat靶细胞的存活(图12e)。
- 还没有人留言评论。精彩留言会获得点赞!