一种地西他滨的合成方法与流程
本发明涉及一种地西他滨的合成方法。
背景技术:
地西他滨是一类β-型异构体,它是经过在母体脱氧核糖支链的异头碳上引入嘧啶类碱基而形成的物质。该药物分别于2006年4月和5月由欧洲emea和美国fda批准上市,用于治疗原发性和继发性骨髓增生异常综合症。据文献报道,地西他滨对镰状细胞性贫血、髓性细胞白血病以及一些实体瘤也有疗效。
地西他滨为目前己知最强的甲基化特异性抑制剂,阻断甲基化可致基因激活与诱导细胞分化。其对细胞毒作用可因胸苷加强,因为胸苷可使本品掺入的量增加,从而增加本品对甲基化的抑制作用。本品为s期细胞周期特异性药物,主要用于骨髓发育不良综合症,包括所有符合法一美一英分类各型且国际预后评分系统为中-1、中-2和高风险组的己经治疗、未经治疗、复发性和继发性脊髓发育不良综合症的患者。
地西他滨经磷酸化后转化为5’-单磷酸脱氧胞苷类似物,在dna聚合酶作用下直接掺入至dna甲基转移酶中,抑制dna的合成及甲基化,从而抑制肿瘤细胞的生长。它在体外不能抑制dna的合成,而在肿瘤细胞内能引发低甲基化,且有维持基因的相关细胞分化和增殖控制功能。由于特殊的药物作用机制,地西他滨引起了广大学者的研究兴趣。核苷通常通过碱基与糖基经糖苷化反应形成糖苷键而制得。地西他滨的合成重点也就在于糖环侧链羟基的保护以及c-n类糖苷键的建立。地西他滨自1964年由pliml和sorm首次合成以来,对其完整的合成路线报道甚少,主要合成方法有经典的silyl-hilbert-johnson法及国内专利cn101307084a等。
silyl-hilbert-johnson法
该方法,在糖环侧链羟基的保护反应中需要把1-o-甲基-2-脱氧-3,4,5-三(-o-苄基)-d-呋喃核糖中的羟甲基进一步转化,延长了实验步骤,使其变的繁琐,使用苄基对羟基进行保护,其产物不易结晶提纯,不利于下步反应和提高最终β-异构体产物的选择性。而且此方法在c-n类糖苷键的建立时使用了重金属盐作为催化剂,容易造成重金属的残留且不易除去。
国内专利cn101307084a
该方法,在糖环侧链羟基的保护反应中也需要把1-o-甲基-2-脱氧-3,4,5-三(-o-苄基)-d-呋喃核糖中的羟甲基进一步转化,延长了实验步骤,使其变的繁琐,使用boc、fmoc或ac等对羟基进行保护,其产物不易结晶提纯,不利于下步反应和提高最终β-异构体产物的选择性,而且不容易脱去保护基团或基团太大从而增大了位阻使产率变低。
因此,亟需一条新的合成方法。
技术实现要素:
本发明的目的在于针对现有技术中地西他滨的合成方法存在步骤长、产物不易提纯和选择性低等缺陷,而提出了一种地西他滨的合成方法。本发明的合成方法具有原料易得,操作安全,条件温和、易控,且酰化保护后得到的固体产物易结晶提纯,有利于下步反应和提高最终β-异构体产物的选择性(ep2048151报道α异构体与β-异构体比例为44.4:55.6,我们是1:2左右),不需要对羟基甲基做进一步的转化,反应步骤得到了简化,各步反应收率好,原子经济性高,适合大量工业化生产等优点。
本发明提供了一种如式i所示的化合物的合成方法,其包括如下步骤:在溶剂中,在tmsotf的作用下,将化合物ii与化合物iii进行如下所示的反应得到化合物i即可;
所述的溶剂可为本领域进行此类反应的常规溶剂,优选为芳烃类、卤代烃类、酯类、酮类和醚类溶剂中的一种或多种。所述的芳烃类溶剂优选为甲苯。所述的卤代烃类溶剂优选为二氯甲烷和/或1,2-二氯乙烷,更优选为1,2-二氯乙烷。所述的酮类溶剂优选为丙酮。所述的醚类溶剂优选为乙醚。所述的酯类溶剂优选为乙酸乙酯。所述的溶剂的用量可为本领域进行此类反应的常规用量,优选其与化合物ii的体积摩尔比为0.5l/mol~10l/mol,更优选为1l/mol~3l/mol,例如:1.85l/mol。
所述的tmsotf的用量可为本领域进行此类反应的常规用量,优选其与化合物ii的摩尔比值为0.5~5,更优选为1~3,例如:2.09。
所述的化合物ii与化合物iii的摩尔比值可为本领域进行此类反应的常规比值,优选为0.5~3,更优选为0.8~1.5,例如:0.98。
所述的反应的温度可为本领域进行此类反应的常规温度,优选为所述的溶剂的回流温度,例如:当所述的溶剂为1,2-二氯乙烷时,所述的反应的温度为80℃~85℃。
所述的反应的进程可以采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行监测,一般以监测到化合物i消失或其含量不再变化时为反应终点,例如,所述的反应的时间为3~6小时。
所述的反应还可进一步包括后处理;所述的后处理方法为此类反应的常规后处理方法,优选包含下列步骤:反应结束后,冷却(例如:冷却至室温),加入弱碱溶液(例如:饱和nahco3溶液)调节ph为中性,有机溶剂(例如:乙酸乙酯)萃取,浓缩去除有机溶剂,加入体积比为1:2的丙酮与水的混合溶剂,加热回流(例如:5小时),搅拌(例如:3小时),过滤,滤饼用体积比为1:2的丙酮与水的混合溶剂洗涤,干燥滤饼,即得化合物i。
所述的如式i所示的化合物的合成方法,还可包括如下步骤:在溶剂中,在碱的作用下,将化合物iv与对氯苯甲酰氯进行如下所示的反应得到化合物ii即可;
在化合物ii的合成方法中,所述的溶剂可为本领域进行此类反应的常规溶剂,优选为卤代烃类、酮类和醚类溶剂中的一种或多种。所述的卤代烃类溶剂优选为二氯甲烷和/或1,2-二氯乙烷,更优选为1,2-二氯乙烷。所述的酮类溶剂优选为丙酮。所述的醚类溶剂优选为乙醚。所述的溶剂的用量可为本领域进行此类反应的常规用量,优选其与化合物iv的体积摩尔比为0.5l/mol~10l/mol,更优选为1l/mol~3l/mol,例如:1.85l/mol。
在化合物ii的合成方法中,所述的碱可为本领域进行此类反应的常用碱,优选为有机碱,更优选为三乙胺。所述的碱的用量可为本领域进行此类反应的常规用量,优选其与化合物iv的摩尔比值为2.2~3.5,例如,2.7。
在化合物ii的合成方法中,所述的对氯苯甲酰氯的用量可为本领域进行此类反应的常规用量,优选其与化合物iv的摩尔比值为2.1~2.8,例如,2.5。
在化合物ii的合成方法中,所述的反应的温度可为本领域进行此类反应的常规温度,优选为所述的溶剂的回流温度,例如:当所述的溶剂为丙酮时,所述的反应的温度为35℃~60℃,例如:55℃。
在化合物ii的合成方法中,所述的反应的进程可以采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行监测,一般以监测到化合物iv消失或其含量不再变化时为反应终点,例如,所述的反应的时间为7~15小时。
在化合物ii的合成方法中,所述的反应还可进一步包括后处理;所述的后处理方法为此类反应的常规后处理方法,优选包含下列步骤:冷却(例如:冷却至室温),加入有机溶剂和水,加入稀酸溶液(例如1m盐酸)调节体系的ph为酸性,有机溶剂萃取,去掉(例如:减压蒸馏)有机溶剂,加入氯仿,过滤,将滤液浓缩得化合物ii。
本发明还提供了一种如下所示的地西他滨的合成方法,其包括如下步骤:a)在溶剂中,在tmsotf的作用下,将化合物ii与化合物iii进行如下所示的反应得到化合物i即可;b)在溶剂中,在碱的作用下,将化合物ii进行如下所示的反应得到地西他滨即可;
步骤a)中,所述的方法和条件均同上所述。
步骤b)中,所述的溶剂可为本领域进行此类反应的常规溶剂,优选为醇类溶剂。所述的醇类溶剂优选为甲醇和/或乙醇。所述的溶剂的用量可为本领域进行此类反应的常规用量,优选其与化合物i的体积摩尔比为0.5l/mol~10l/mol,更优选为1l/mol~4l/mol,例如:2.0l/mol。
步骤b)中,所述的碱可为本领域进行此类反应的常规碱,优选为有机碱,更优选为甲醇钠和/或乙醇钠。所述的碱的用量可为本领域进行此类反应的常规用量,优选其与化合物i的摩尔比值为0.5~5,更优选为1~3,例如:1.48。
步骤b)中,所述的反应的温度可为本领域进行此类反应的常规温度,优选为60℃~80℃。
步骤b)中,所述的反应的进程可以采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行监测,一般以监测到化合物i消失或其含量不再变化时为反应终点,例如,所述的反应的时间为5~7小时。
步骤b)中,所述的反应还可进一步包括后处理;所述的后处理方法为此类反应的常规后处理方法,优选包含下列步骤:反应结束后,降温(例如:50℃),加入阳离子树脂调节ph<3.5,趁热过滤,用甲醇洗涤树脂,合并洗脱液和滤液,除去(例如:减压蒸馏)溶剂,得到浆料,向其中加入丙酮,加热回流,降温,过滤,滤饼用丙酮洗涤,真空干燥得地西他滨。
所述的化合物iii和iv的合成方法可以参考国内专利cn101307084a中相应化合物的合成方法。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明中,如未限定操作温度,均在室温下进行。所述的室温为0℃~35℃,优选20℃~30℃。
本发明的积极进步效果在于:所用的合成方法操作安全,条件温和、易控,且酰化保护后得到的固体产物易结晶提纯,有利于下步反应和提高最终β-异构体产物的选择性,不需要对羟基甲基做进一步的转化,反应步骤得到了简化,各步反应收率好,原子经济性高,适合大量工业化生产。
附图说明
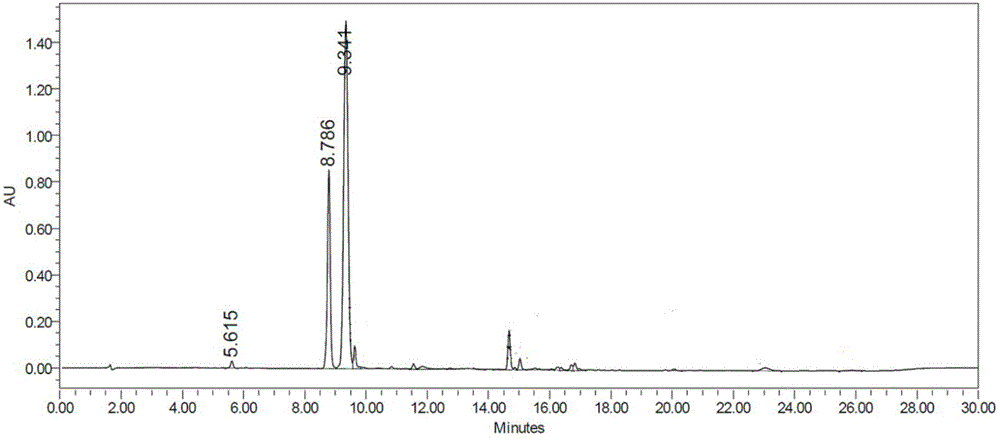
图1为实施例4中所得产物的α异构体与β-异构体的hplc图谱。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例11-o-甲基-2-脱氧-d-呋喃核糖(化合物iv)的制备
5l的反应瓶中,加入2l的1%的hcl甲醇溶液,冷却至0~5℃,加入2-脱氧-d-核糖100克(755.55mmol),自然恢复至室温,反应1~2小时后,tlc检测原料2-脱氧-d-核糖反应完全(二氯甲烷:甲醇=8:1v/v)。滴加入饱和碳酸氢钠溶液约30ml,调节ph=8左右,过滤减压浓缩溶剂,加入200的甲苯,减压浓缩干,继续加入200的甲苯,减压浓缩干得108克浆状液体(收率:98%),直接用于下一步。
实施例21-o-甲基-2-脱氧-3,5-双(-o-对氯苯甲酰)-d-呋喃核糖(化合物ii)的制备
5l的反应瓶中,加入上一步所得的1-o-甲基-2-脱氧-d-呋喃核糖的浆状液体108克(728.94mmol),加入丙酮2l,室温下滴加对氯苯甲酰氯318.9克(1822.35mmol),滴加完之后继续滴加三乙胺199.1克(1968.14mmol),滴完之后升温55℃回流10小时,tlc检测原料1-o-甲基-2-脱氧-d-呋喃核糖反应完全(二氯甲烷:甲醇=8:1v/v)。体系过滤,滤液浓缩干,加入450ml的二氯甲烷和2550ml的水,慢慢加入冷的1%的盐酸水溶液200ml调节ph为酸性,萃取分层,有几层浓缩去二氯甲烷,加入500ml的氯仿搅拌,过滤,弃去滤渣,浓缩干得303.8克(收率:98%),纯度:96.3%。
实施例32,4-二(三甲基硅氧基)-5-氮杂胞嘧啶(化合物iii)的制备
5-氮杂胞嘧啶81.79克(729.69mmol),六甲基二硅胺烷800ml,硫酸铵3.37克(25.51mmol)加入2升的反应瓶中,氮气保护下加热回流至澄清,继续回流2.5h。减压蒸出过量的六甲基二硅胺烷,再加入两次1l的甲苯进行减压蒸馏带出残余的六甲基二硅胺烷,得得2,4-二(三甲基硅氧基)-5-氮杂胞嘧啶,待用。
实施例41-(3,5-二-o-对氯苯酰基-2-脱氧-d-核糖)-5-氮杂胞嘧啶(化合物i)的制备
将实施例3所得的2,4-二(三甲基硅氧基)-5-氮杂胞嘧啶溶解于1l的二氯乙烷中,加入5l的反应瓶中,再把实施例2所得1-o-甲基-2-脱氧-3,5-双(-o-对氯苯甲酰)-d-呋喃核糖303.8克(714.38mmol)溶解于1l的二氯乙烷中加入该反应瓶中,室温下缓慢滴加tmsotf332.3克(1495.09mmol)的300ml的二氯乙烷溶液,滴加完毕后升温至85℃回流5小时,tlc检测原料1-o-甲基-2-脱氧-3,5-双(-o-对氯苯甲酰)-d-呋喃核糖反应完全(石油醚:乙酸乙酯=3:1v/v)。冷至室温,加入1.5l的饱和碳酸氢钠调ph为中性,搅拌后静置分层,萃取后收集有机层,旋蒸浓缩去溶剂,加入500ml的丙酮、1l的水,升温至80℃回流5小时,冷却至0℃,搅拌3小时,过滤,滤饼用丙酮:水=1:2v/v的溶液200ml洗涤两到三次,60℃下真空干燥,即得1-(3,5-二-o-对氯苯酰基-2-脱氧-d-核糖)-5-氮杂胞嘧啶126.35克(250.03mmol),收率35%,纯度:95.1%,图1为实施例4中所得产物的α异构体与β-异构体的hplc图谱,二者比例为1:2左右。
实施例54-氨基-1-(2-脱氧-β-d-赤型-呋喃核糖)-1,3,5-三嗪-2-酮(地西他滨)的制备
在氩气保护下,1l的反应瓶中,将实施例4所得的1-(3,5-二-o-对氯苯酰基-2-脱氧-d-核糖)-5-氮杂胞嘧啶126.35克(250.03mmol)溶于含20克甲醇钠的500ml的甲醇中,升温至65℃回流6小时,反应结束后,降温至50℃,加入阳离子树脂调节ph<3.5,称热过滤,并用适量甲醇洗涤树脂,洗脱液和滤液合并,然后进行旋转蒸发除去甲醇溶剂,往得到的浆料中加入200ml的丙酮,加热至70℃回流3小时,然后降温至10℃并保温30min~1h,过滤,滤饼用50ml的丙酮洗涤两次,然后再真空干燥箱中干燥得最终产品4-氨基-1-(2-脱氧-β-d-赤型-呋喃核糖)-1,3,5-三嗪-2-酮(地西他滨)34.24克(150.02mmol)。纯度:99.9%,熔点:191.6~192.1℃,核磁:hnmr(300mhz,dmso-d6)δ2.16(2h,m),3.61(2h,m),3.81(1h,d,j=3.0hz),4.24(1h,brs),5.05(1h,t,j=5.1hz),5.24(1h,d,j=4.2hz),6.03(1h,t,j=6.3hz),7.50(1h,s)7.53(1h,s),8.51(1h,s).
实施例62,4-二(三甲基硅氧基)-5-氮杂胞嘧啶(化合物iii)的制备
5-氮杂胞嘧啶80克(713.72mmol),六甲基二硅胺烷800ml,硫酸铵3.30克(24.95mmol)加入2升的反应瓶中,氮气保护下加热回流至澄清,继续回流2.5h。减压蒸出过量的六甲基二硅胺烷,再加入两次1l的甲苯进行减压蒸馏带出残余的六甲基二硅胺烷,得得2,4-二(三甲基硅氧基)-5-氮杂胞嘧啶,待用。
实施例71-(3,5-二-o-对氯苯酰基-2-脱氧-d-核糖)-5-氮杂胞嘧啶(化合物i)的制备
将实施例3所得的2,4-二(三甲基硅氧基)-5-氮杂胞嘧啶溶解于800ml的甲苯中,加入5l的反应瓶中,再把1-o-甲基-2-脱氧-3,5-双(-o-对氯苯甲酰)-d-呋喃核糖364.22克(856.46mmol)溶解于800ml的甲苯中加入该反应瓶中,室温下缓慢滴加tmsotf228.4克(1027.75mmol)的300ml的甲苯溶液,滴加完毕后升温至110℃回流5小时,tlc检测原料1-o-甲基-2-脱氧-3,5-双(-o-对氯苯甲酰)-d-呋喃核糖反应完全(石油醚:乙酸乙酯=3:1v/v)。冷至室温,加入1.5l的饱和碳酸氢钠调ph为中性,搅拌后静置分层,萃取后收集有机层,旋蒸浓缩去溶剂,加入500ml的丙酮、1l的水,升温至80℃回流5小时,冷却至0℃,搅拌3小时,过滤,滤饼用丙酮:水=1:2v/v的溶液200ml洗涤两到三次,60℃下真空干燥,即得1-(3,5-二-o-对氯苯酰基-2-脱氧-d-核糖)-5-氮杂胞嘧啶138.5克(274.06mmol),收率32%,纯度:94.8%。
- 还没有人留言评论。精彩留言会获得点赞!