一种耐高温Cas蛋白的用途及靶标核酸分子的检测方法和试剂盒与流程
本发明属于生物技术领域,具体地说,本发明涉及耐高温cas蛋白的用途及靶标核酸分子的检测方法和试剂盒。
背景技术:
快速核酸分子检测是公众健康、环境检测、刑侦等领域非常关键的技术。核酸分子检测,不仅可以用于检测人、动植物等是否感染了病原体,也可以检测如遗传病或癌症风险检测、个人用药提供辅助参考、水体是否有微生物污染等。目前,已经开发出了不少核酸检测方法,如realtimepcr、fish杂交技术(fluorescenceinsituhybridization)等。但是,仍需要开发兼有快速、廉价、灵敏的核酸检测技术。
近几年来,crispr技术在基因组编辑领域表现出了巨大的应用价值。完成这一任务的主要是cas9等dna(或rna)靶向的核酸内切酶。人们只要通过简单的设计向导rna,就可以将cas9等蛋白靶向到任意含pam(如cas9的pam序列为ngg)的核酸靶标序列,并引起双链dna的断裂(或rna的切割)。
除了基因组编辑,crispr还可以用于基因调控、表观遗传学编辑、功能基因筛选、基因组成像等。最近,crispr这个丰富的工具箱开始在核酸检测领域出现并受到关注。
首先出现的crispr相关核酸检测技术是基于dcas9(deadcas9)蛋白的技术。dcas9是将cas9的两个催化切割活性位点突变,从而失去切割双链的活性,但dcas9仍然保留由向导rna引导并结合特定dna的能力。chen等利用dcas9与egfp荧光蛋白融合,可以利用特定向导rna引导,通过显微镜可以观察到靶标序列处的荧光。2017年,guk等报导了基于dcas9/sgrna-sgi的dna-fish系统,可以选择性检测甲氧西林耐受的金黄色葡萄球菌。
2013年,collins团队建立了在纸片上即可检测寨卡(zika)病毒的方法。该方法涉及到rna的提取、扩增和toehold反应检测过程,通过设计,如果一种病毒株恰巧与另一株在pam的位点序列处不同(即一种是ngg,另一种则不同),即可通过cas9靶向切割含pam的菌株,从而影响扩增过程和toehold的检测。
最近,另一种基于cas9的核酸检测方法的原理是crispr/cas9触发的等温扩增方法,也可用于位点特异性核酸检测。这个技术涉及到crispr/cas9的切割、切口酶和dna聚合酶,这种方法不仅可以检测到0.82amol的靶标浓度,而且可以区分核酸分子单个碱基的区别。该方法的靶标区分相对前一种方法来说更加普遍适用。
另一种核酸检测的原理则是基于一些cas蛋白具有的“反式切割”(或称“旁路切割”)效应。2016年,张锋等发现cas13a(旧称c2c2)具有旁路切割活性,即当cas13a结合靶标rna序列之后,会表现出杂乱切割其他rna的特性,从而用于特异性核酸检测,称为sherlock(specifichighsensitivityenzymaticreporterunlocking)技术。
虽然已有不少crispr相关的核酸检测方法,然而本领域仍需要开发更快、更简便、更便宜的核酸检测方法。
技术实现要素:
本发明的目的就是开发一种crispr相关的更快、更简便、更便宜的核酸检测方法。
本发明的第一方面,提供了一种用于检测靶标核酸分子的检测体系,所述体系包含:
(a)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
(b)向导rna,所述向导rna引导cas12b蛋白特异性结合于靶标核酸分子;和
(c)核酸探针,所述核酸探针为单链dna;
其中,所述的靶标核酸分子为靶标dna。
在另一优选例中,所述的检测包括:定性检测或定量检测。
在另一优选例中,所述的检测体系还含有(d)缓冲液。
在另一优选例中,所述的检测体系还含有待检测的靶标核酸分子。
在另一优选例中,所述的检测体系还含有:
(e1)用于扩增靶标dna的聚合酶;
(e2)任选的用于反转录的反转录酶;
(e3)用于扩增反应和/或反转录反应的dntp。
在另一优选例中,所述的检测体系还含有用于lamp反应的试剂。
在另一优选例中,所述的待检测的靶标核酸分子在所述检测体系中的浓度为1×10-9nm-1×103nm;较佳地1×10-8nm-1×102nm。
在另一优选例中,所述的待检测的靶标核酸分子在所述检测体系中的浓度为1-100拷贝/微升或1-1×1015拷贝/微升,较佳地1-10拷贝/微升,更佳地1-5拷贝/微升。
在另一优选例中,所述的检测体系中,所述核酸探针与所述靶标核酸分子的摩尔比为103:1至1014:1,较佳地104:1至107:1。
在另一优选例中,所述的向导rna的长度为16-25nt,较佳地16-22nt,更佳地16-20nt。
在另一优选例中,所述的向导rna的长度为18-25nt,较佳地18-22nt,更佳地18-20nt。
在另一优选例中,当靶标核酸分子为ssdna时,所述的靶标核酸分子的检测位点位于所述向导rna的pam序列下游第9位或第10-16位,较佳地10-14位,更佳地10-12位。
在另一优选例中,当所述的检测位点位于所述向导rna的pam序列下游第9位,所述的检测位点为g。
在另一优选例中,所述的检测位点位于所述向导rna的pam序列下游9-20位,较佳地10-18位,更佳地10-16位。
在另一优选例中,所述的靶标核酸分子的检测位点位于所述向导rna的pam序列下游第1-12位,更佳地位于第1,3,10位。
在另一优选例中,所述的向导rna的长度为15-30nt,较佳地15-18nt。
在另一优选例中,所述的靶标dna包括基于rna反转录形成的dna。
在另一优选例中,所述的靶标dna包括cdna。
在另一优选例中,所述的靶标dna选自下组:单链dna、双链dna、或其组合。
在另一优选例中,所述的核酸探针带有荧光基团和淬灭基团。
在另一优选例中,所述的荧光基团和淬灭基团各自独立地位于所述核酸探针的5'端、3'端和中部。
在另一优选例中,所述的核酸探针的长度为3-300nt,较佳地5-100nt,更佳地6-50nt,最佳地8-20nt。
在另一优选例中,所述靶标核酸分子包括来源于选自下组的靶标核酸分子:植物、动物、昆虫、微生物、病毒、或其组合。
在另一优选例中,所述的靶标dna是人工合成或天然存在的dna。
在另一优选例中,所述的靶标dna包括野生型或突变型的dna。
在另一优选例中,所述的靶标dna包括由rna逆转录或扩增而获得的dna,如cdna等。
在另一优选例中,所述的cas12b蛋白选自下组:aaccas12b(alicyclobacillusacidoterrestris)、aac2cas12b(alicyclobacillusacidiphilus)、akacas12b(alicyclobacilluskakegawensis)、amacas12b(alicyclobacillusmacrosporangiidus)、ahecas12b(alicyclobacillusherbarius)、acocas12b(alicyclobacilluscontaminans)。
在另一优选例中,所述的核酸探针包括带有可检测标记的单链dna。
在另一优选例中,所述的单链dna为荧光和生物素标记的单链dna。
在另一优选例中,所述的单链dna为荧光标记的单链dna。
在另一优选例中,所述的单链dna为在5'端标记荧光基团hex并在3'端标记淬灭基团bhq1后的荧光探针。
在本发明的第二方面,提供了一种用于检测snp(单核苷酸多态性)或核苷酸突变(包括单碱基或多碱基突变)的检测体系,所述体系包含第一检测体系和第二检测体系;
其中第一检测体系包括:
(a1)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
(b1)第一向导rna,所述第一向导rna引导cas12b蛋白特异性结合于靶标核酸分子;和
(c1)核酸探针,所述核酸探针为单链dna;
第二检测体系包括:
(a2)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
(b2)第二向导rna,所述第二向导rna引导cas12b蛋白特异性结合于靶标核酸分子;和
(c2)核酸探针,所述核酸探针为单链dna;
其中,所述的靶标核酸分子为靶标dna;
并且,所述的第一向导rna和第二向导rna针对包含所述snp位点的同一核酸序列区域,且第一向导rna针对该snp位点的野生型(或未突变的)核酸序列,而所述第二向导rna针对该snp位点的突变型核酸序列。
在另一优选例中,所述体系还包括一空白对照体系。
在另一优选例中,所述空白对照体系包括:
(a3)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;和
(c3)核酸探针,所述核酸探针为单链dna。
在另一优选例中,所述snp位点位于所述向导rna的pam序列下游第9位或第10-16位,较佳地10-14位,更佳地10-12位。
在另一优选例中,所述snp位点位于所述向导rna的pam序列下游第9位,所述的检测位点为g。
在另一优选例中,所述snp位点位于所述向导rna的pam序列下游9-20位,较佳地10-18位,更佳地10-16位。
在另一优选例中,所述的第一或第二向导rna的长度为16-25nt,较佳地16-22nt,更佳地16-20nt。
在另一优选例中,所述的第一或第二向导rna的长度为18-25nt,较佳地18-22nt,更佳地18-20nt。
在本发明的第三方面,提供了一种用于检测靶标核酸分子的试剂盒,所述试剂盒包括:
i)第一容器以及位于第一容器内的cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
ii)任选的第二容器以及位于第二容器内的向导rna,所述向导rna引导所述cas蛋白特异性结合于靶标核酸分子;
iii)第三容器以及位于第三容器内的核酸探针;
iv)任选的第四容器以及位于第四容器内的缓冲液;
其中,所述的靶标核酸分子为靶标dna。
在另一优选例中,所述的第一容器、第二容器、第三容器和第四容器中的任何二个、三个、或四个(或全部)可以是相同或不同容器。
在另一优选例中,所述的核酸探针带有荧光基团和淬灭基团。
在另一优选例中,所述试剂盒还包括缓冲液。
在另一优选例中,所述试剂盒还包括:
v)第五容器以及位于第五容器内的用于扩增靶标dna的聚合酶;
vi)任选的第六容器以及位于第六容器内的用于反转录的反转录酶;
vii)第七容器以及位于第七容器内的用于扩增反应和/或反转录反应的dntp。
在另一优选例中,所述的检测体系还含有用于lamp反应的试剂。
在另一优选例中,所述第五容器、第六容器和第七容器可以是相同容器或不同容器。
在另一优选例中,所述第一容器至第七容器中的二个、多个或全部可以是相同容器或不同容器。
在本发明的第四方面,提供了一种检测样本中是否存在靶标核酸分子的方法,包括以下步骤:
(i)提供本发明第一方面所述的用于检测靶标核酸分子的检测体系,并且所述的检测体系还含有待检测的样本;和
(ii)检测所述检测体系中的核酸探针是否被cas12b蛋白进行切割,所述的切割为旁路单链dna的反式切割;
其中,所述核酸探针被cas12b蛋白切割,则表示所述样本中存在靶标核酸分子;而所述核酸探针不被cas12b蛋白切割,则表示所述样本中不存在靶标核酸分子。
在另一优选例中,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白。
在另一优选例中,所述的待检测的样本包括未经扩增的样本以及经过扩增(或核酸扩增)的样本。
在另一优选例中,所述的待检测的样本是经过扩增而获得的样本。
在另一优选例中,所述核酸扩增的方法选自下组:pcr扩增、lamp扩增、rpa扩增、连接酶链式反应、分支dna扩增、nasba、sda、转录介导扩增、滚环扩增、hda、spia、near、tma和smap2。
在另一优选例中,所述的pcr包括高温pcr、常温pcr、或低温pcr。
在另一优选例中,所述方法用于检测靶位点处的核酸是否在snp、点突变、缺失、和/或插入。
在另一优选例中,当所述的靶位点的上下游(-20nt至+20nt范围内,,更佳地-16nt至+16nt范围内)缺乏pam序列时,采用引入pam的引物进行核酸扩增。
在另一优选例中,所述的引入pam的引物具有从5'-3'的式i结构:
p1-p2-p3(i)
式中,
p1为位于5'端的与靶标核酸分子的序列互补或非互补的5'区段序列;
p2为pam序列;
p3为位于3'端的与靶标核酸分子的序列互补3'区段序列。
在另一优选例中,所述的pam引物特异性结合于靶标核酸分子的上游或下游。
在另一优选例中,p1的长度为0-20nt。
在另一优选例中,p3的长度为5-20nt。
在另一优选例中,所述的pam引物的长度为16-50nt,较佳地20-35nt。
在另一优选例中,所述的互补包括完全互补和部分互补。
在另一优选例中,所述的核酸扩增中使用至少一条引物含有pam序列。
在另一优选例中,当所述的靶位点的上下游(-20nt至+20nt范围内,较佳地-15nt至+15nt范围内,更佳地-10nt至+10nt范围内)含有pam序列时,则可采用含有或不含有pam序列的引物,并且扩增出的扩增产物含有所述pam序列。
在另一优选例中,在步骤(ii)中的检测包括荧光检测法。
在另一优选例中,所述荧光检测法采用酶标仪或者荧光分光光度计进行检测。
在本发明的第五方面,提供了一种检测样本中是否存在靶标核酸分子的方法,包括以下步骤:
(i)提供一检测体系,所述检测体系包括:
(a)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
(b)向导rna,所述向导rna引导cas12b蛋白特异性结合于靶标核酸分子;
(c)核酸探针,所述核酸探针为单链dna;
其中,所述的靶标核酸分子为靶标dna;
(d)缓冲液
(e1)用于扩增靶标dna的聚合酶;
(e2)任选的用于反转录的反转录酶;
(e3)用于扩增反应和/或反转录反应的dntp;和
(f)待检测的检测样本;
(ii)对所述检测体系进行反转录和/或扩增反应,从而获得经反转录和/或经扩增的检测体系;
(iii)检测上一步骤中的所述检测体系中的核酸探针是否被cas12b蛋白进行切割,所述的切割为旁路单链dna的反式切割;
其中,所述核酸探针被cas12b蛋白切割,则表示所述样本中存在靶标核酸分子;而所述核酸探针不被cas12b蛋白切割,则表示所述样本中不存在靶标核酸分子。
在另一优选例中,所述的步骤(ii)中,维持所述检测体系的温度在50-70℃,较佳地50-65℃,更佳地55-65℃。
在另一优选例中,所述的步骤(iii)中,维持所述检测体系的温度在25-70℃,较佳地48-65℃。
在另一优选例中,所述核酸扩增的方法选自下组:pcr扩增、lamp扩增、rpa扩增、连接酶链式反应、分支dna扩增、nasba、sda、转录介导扩增、滚环扩增、hda、spia、near、tma和smap2。
在另一优选例中,所述的核酸扩增包括:lamp扩增。
在另一优选例中,所述检测体系中,待检测的检测样本的核酸浓度为1×10-11-1×10-5nm,较佳地1×10-9-1×10-6nm,更佳地1×10-8-1×10-7nm。
在另一优选例中,所述的检测包括:定性检测或定量检测。
在另一优选例中,所述定量检测为绝对定量检测(例如与数字pcr技术相结合进行定量检测)。
在另一优选例中,所述步骤(ii)和(iii)的总时间≤2小时,较佳地≤1.5小时,更佳地≤1小时(如30-60分钟)。
在本发明的第六方面,提供了一种cas12b蛋白的用途,所述cas12b蛋白被用于制备基于旁路单链dna切割检测靶标核酸分子的检测试剂或试剂盒,其中所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白。
在另一优选例中,所述的cas12b蛋白选自下组:aaccas12b(alicyclobacillusacidoterrestris)、aac2cas12b(alicyclobacillusacidiphilus)、akacas12b(alicyclobacilluskakegawensis)、amacas12b(alicyclobacillusmacrosporangiidus)、ahecas12b(alicyclobacillusherbarius)、acocas12b(alicyclobacilluscontaminans)。
在本发明的第七方面,提供了一种用于检测样本中是否存在靶标核酸分子的设备,所述设备包括:
(a)扩增反应-旁路切割反应模块,所述模块用于对数字化的反应体系进行核酸扩增反应和旁路切割反应,其中所述的旁路切割反应是cas12b蛋白介导的进行旁路切割;和(b)信号检测模块,所述模块用于检测各数字化的反应体系是否发生了cas12b蛋白介导的进行旁路切割。
在另一优选例中,所述的核酸扩增反应和旁路切割反应是同时进行的。
在另一优选例中,所述的扩增反应-旁路切割反应模块还包括一温控单元,所述温控单元用于将位于所述设备中的数字化的反应体系的温度扩增在预定的温度。
在另一优选例中,所述的预定温度是50-70℃,较佳地50-65℃,更佳地55-65℃。
在另一优选例中,所述的预定温度在25-70℃,较佳地48-65℃。
在另一优选例中,所述整个扩增反应和旁路切割反应过程中,预定温度是基本相同的。
在另一优选例中,所述的预定温度的波动范围在±5℃之内,较佳地±3℃之内,更佳地±1℃之内。
在另一优选例中,所述的核酸扩增为等温扩增,或变性-复性-延伸温度的差异≤10℃(较佳地≤5℃,更佳地≤3℃)的扩增。
在另一优选例中,所述的核酸扩增包括:lamp扩增。
在另一优选例中,所述的数字化的反应体系包括微滴式数字pcr(ddpcr)反应体系或芯片数字pcr(cdpcr)反应体系。
在另一优选例中,所述的数字化的反应体系包括多个独立的反应体系单元,每个反应体系单元均包括:
(a)cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
(b)向导rna,所述向导rna引导cas12b蛋白特异性结合于靶标核酸分子;
(c)核酸探针,所述核酸探针为单链dna;
其中,所述的靶标核酸分子为靶标dna;
(d)缓冲液
(e1)用于扩增靶标dna的聚合酶;
(e2)任选的用于反转录的反转录酶;
(e3)用于扩增反应和/或反转录反应的dntp;和
(f)待检测的检测样本,
其中,由于稀释化处理(即数字化处理),每个独立的反应体系中或者含有1个拷贝的来自检测样本的核酸,或者含有0个拷贝的来自检测样本的核酸。
在另一优选例中,每个反应体系单元是相同的,除了各反应体系单元含有1个或0个拷贝的来自检测样本的核酸之外。
在另一优选例中,所述的反应体系单元是微滴。
在另一优选例中,所述的反应体系单元是位于芯片的微孔中的微反应体系。
在另一优选例中,所述的设备还包括加样模块、和/或控制模块。
在另一优选例中,所述的信号检测模块包括成像模块。
在另一优选例中,所述的成像模块包括一荧光检测单元。
在另一优选例中,所述荧光检测单元通过照射所述数字化的反应体系,从而激发所述反应体系产生荧光信号,并将所述荧光信号转换成数字信号,并优选地发送至所述控制模块。
在另一优选例中,所述控制模块基于所述荧光信号(优选地,该荧光信号已转换成的数字信号)的数量和所述反应体系单元的总数量进行运算分析,从而得到靶核酸的定量检测结果(如浓度或拷贝数)。
在另一优选例中,所述的设备是数字pcr检测装置。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
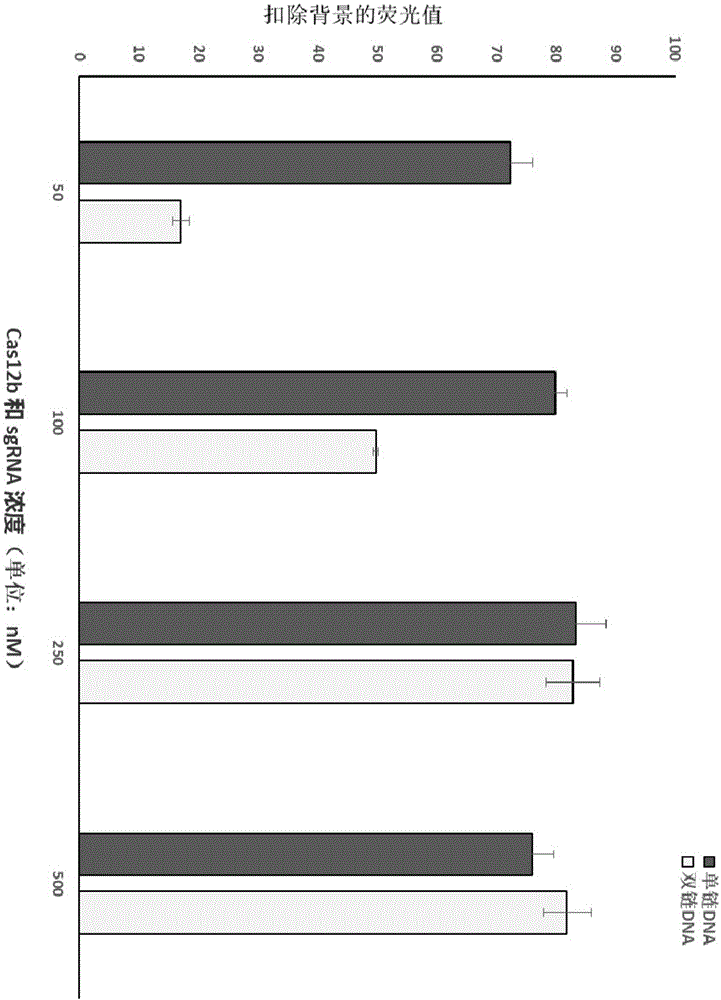
图1显示了分别dsdna和ssdna作为靶标dna情况下,不同浓度的cas12b和sgrna进行trans切割探针产生的荧光强度。
图2显示了分别dsdna和ssdna作为靶标dna情况下,pam序列处不同碱基对trans切割荧光检测强度的影响。
图3显示了ssdna或dsdna作为靶标时,cas12b的trans切割速率。
图4显示了不同温度条件下,以dsdna或ssdna为靶标cas12b的trans切割荧光强度。
图5显示了通过梯度稀释靶标序列,cas12b以dsdna或ssdna为靶标的检测灵敏度(即trans切割的荧光强度)。
图6显示了cas12b结合lamp扩增反应(即holmesv2.0方法)对靶标的检测灵敏度。
图7利用不同长度引导序列的sgrna,分别测定ssdna靶标序列的1-12位中的单碱基突变对cas12btrans切割荧光强度的影响。
图8利用引导序列为16ntsgrna,分别测定ssdna靶标序列的1-16位中的单碱基突变对cas12btrans切割荧光强度的影响。
图9利用引导序列为16ntsgrna,分别测定ssdna靶标序列的1-16位中的单碱基突变成任意其他3种碱基对cas12btrans切割荧光强度的影响。
图10利用引导序列为16ntsgrna,分别测定3种不同ssdna靶标序列(rs5082,rs1467558,rs2952768)的8-16位中的单碱基突对cas12btrans切割荧光强度的影响。
图11利用不同长度引导序列的sgrna,测定dsdna靶标序列的1-16位中任意单碱基突变对cas12btrans切割荧光强度的影响。
图12显示了在50℃、55℃、60℃、65℃条件下,将lamp与cas12b检测实现一步法反应。
图13holmesv2.0(lamp结合cas12b)测试大肠杆菌的gyrb位点,鉴定是否存在大肠杆菌以及通过梯度稀释测试检测灵敏度。(经过涂板测试,0.005od的大肠杆菌大约是7000个)
图14holmesv2.0(lamp结合cas12b)测试y染色体sry位点,利用唾液样本鉴定性别(dnmt1-3位点是染色体上的基因位点,为正对照)。
图15非对称或对称pcr扩增结合cas12b检测snp位点(rs5082)。可以看出,由于rs5082位点附近没有pam序列,因此产生单链dna的非对称pcr可以更明显的区分snp位点。
图16通过设计引物,通过lamp扩增引入pam序列后,结合cas12b检测snp位点(rs5082)。可以看出,由于rs5082位点附近没有pam序列,因此产生加入pam的lamp扩增方法(引物中带pam序列),可以更好地区分snp位点。
图17holmesv2.0(lamp结合cas12b)对rna病毒jev(japaneseencephalitisvirus;日本乙型脑炎病毒)的检测。
图18holmesv2.0(lamp结合cas12b)一步法定量微量dna。通过稀释不同浓度的模板dna,用lamp-cas12b一步法在荧光定量仪中实时检测荧光值(55℃反应)。
图19以荧光值达到600,000的时间点为y轴(采用图18中的数据),以浓度的lg绝对值为x轴作图,得到近似直线的趋势线。可以以此方程进行靶标序列的定量。
图20数字holmes(lamp结合cas12b与clarity芯片)方法,对核酸样品的绝对定量检测。
具体实施方式
本发明人通过广泛而深入的研究,通过对cas12b的切割特性的研究,首次开发了一种靶标核酸检测的技术方案。实验结果表明,采用本发明技术方案可快速、高灵敏和高准确地检测微量核酸,可应用于例如唾液样品快速鉴定性别、大肠杆菌污染检测、snp基因型的快速鉴定、rna病毒检测以及微量dna样品浓度测定等不同领域。在此基础上完成了本发明。
术语
术语“向导rna”或“grna”或“sgrna”是指引导cas蛋白(如cas12b蛋白)特异性结合靶标dna序列的rna。
术语“crispr”是指成簇的、规律间隔的短回文重复序列(clusteredregularlyinterspacedshortpalindromicrepeats),该序列是许多原核生物的免疫系统。
术语“cas蛋白”是指crispr-associated蛋白,它是crispr系统中的相关蛋白。
术语“cas12a”(旧称“cpf1”)是指crrna依赖的内切酶,它是crispr系统分类中v-a型的酶。
术语“cas12b”,“c2c1”可互换使用,是指sgrna依赖的内切酶,它是crispr系统分类中v-b型的酶。
术语“lamp”是环介导等温扩增技术(loop-mediatedisothermalamplification),是一种适用于基因诊断的恒温核酸扩增技术。
术语“pam”是指前间区序列邻近基序(protospacer-adjacentmotif),是cas12b切割双链dna所必须,aaccas12b的pam为ttn序列。
术语“pcr”是指“聚合酶链式反应”,用于大量扩增目的dna片段的一种方法。
holmesv2.0是指一小时、低价、多用途、高效系统版本2.0(one-hourlow-costmultipurposehighlyefficientsystemversion2.0),即是基于cas12b的核酸检测方法。
检测方法
本发明公开了一种靶标核酸分子的检测方法,将含有待检靶标核酸分子的反应体系中,向导rna、cas蛋白、核酸探针、缓冲液,然后对其进行荧光检测。
本发明提供了一种高特异性地快速检测靶标核酸分子的检测方法。一旦靶标dna(单链或者双链)、sgrna和cas12b蛋白形成三元复合体时,该复合物会切割体系中其他的单链dna分子。
通过设计sgrna靶向靶标dna(需要检测的一段dna序列);向检测体系中加入sgrna和cas12b蛋白;当靶标dna存在时(单链dna或双链dna),cas12b与sgrna以及靶标dna形成三元复合体,同时该复合物行使其旁路切割的活性并切割带荧光信号标记的核酸探针,从而发出荧光。。
代表性的核酸探针是单链dna,其两端分别连有发光基团和淬灭基团,因此一旦该探针被切断后,发光基团可以发光。
在本发明中,通过检测荧光即可得知待检测体系中是否含有靶标dna分子。
在本发明中,合适的cas蛋白为cas12b,较佳地所述的cas12b优选为aaccas12b(alicyclobacillusacidoterrestris)、aac2cas12b(alicyclobacillusacidiphilus)、akacas12b(alicyclobacilluskakegawensis)、amacas12b(alicyclobacillusmacrosporangiidus)、ahecas12b(alicyclobacillusherbarius)、acocas12b(alicyclobacilluscontaminans)。
待检靶标核酸分子的反应体系中的待检靶标核酸分子可以是未经扩增的,或经过扩增后得到的,和/或经过反转录扩增。
本发明检测方法可检测不同物种的核酸分子,例如哺乳动物、植物、或微生物、病毒的核酸分子。本发明方法特别适合用于检测病原微生物、基因突变或特异靶标dna或rna。
使用本发明的方法可快速检测样品中是否含有特异dna序列。此外,通过与扩增技术的结合(如lamp、pcr、非对称pcr、rpa等),该检测方法的灵敏度可以得到大幅度提高。
优选地,可将cas12b的旁路切割与核酸扩增(如等温扩增、lamp扩增)以及其他技术(如数字pcr)等进行结合。
在本发明的一个优选例中,当将基于cas12b的旁路切割的检测与核酸扩增结合时,可将检测灵敏度提高到10-8nm或更低浓度。
此外,当将基于cas12b的旁路切割的检测与数字pcr结合时,可将检测灵敏度提高到1拷贝/1个反应体系(如一个微液滴),从而几乎可以满足所有的检测灵敏度的需要。
基于cas12b的核酸检测方法和holmesv2.0(one-hourlow-costmultipurposehighlyefficientsystemversion2.0)
核酸探测方法的建立
利用cas12b的特性,本发明开发了特异性检测核酸分子的方法,称之为holmesv2.0(one-hourlow-costmultipurposehighlyefficientsystemversion2.0)。正如该技术的名称一样,其特点是一小时、低成本、多用途、高效率、很简便的测试法。
在整个反应体系中,可分为两个大的步骤,一个是对模板核酸的扩增,另一个是cas12b蛋白的特异性核酸检测;或者也可以将这两步结合成一步。
在本发明中,在一优选例中对于核酸的扩增运用了lamp的方法或者非对称pcr的方法,但实际上,任何的扩增方法都可结合第二步的核酸检测,例如等温扩增方法rpa等。
初始的核酸不限于双链dna,也可以是单链dna或rna,因此本方法适用于多种类型的核酸分子。
对于核酸检测阶段,其中3个组分是实验的关键,分别为cas12b,sgrna和核酸探针。除了实施例中提到的aaccas12b,其他cas12b蛋白同样适用于该方法。此外,其它类型的cas蛋白(如cas12c蛋白)也适用于本发明保护的范畴。
对于作为引导作用的sgrna,经过人工修饰等改造后,在体系中会更加稳定。在核酸探针的选择上,本发明选用了hex和bhq1标记的短单链dna(也用了fam和eclipse标记),其他可检测的任何标记方式理论上都是适用的,只要该核酸探针被切割后产生可检测的差异。或者,核酸探针也可以设计成可以跟化合物结合后发荧光,从而探测是否该探针被切断。
为了便于理解,进一步对本发明基于cas12b的核酸检测方法(包括优选的“holmes”v2.0方法)的各种不同特性进行描述。
cas12btrans切割(反式切割)活性鉴定:虽然在序列上,cas12a和cas12b的差异较大,但本发明人实验结果表明,cas12b也具有trans切割活性。
首先,将旁路dna设计成荧光探针,其组成是12nt的随机序列,并在5'端标记荧光基团hex,在3端标记淬灭基团bhq1(hex-n12-bhq1)。当体系中含有标靶dna片段时(dsdna或ssdna),即形成靶标dna、sgrna和cas12b蛋白的三元复合体,此时该探针即被切割,同时通过荧光检测仪的检测hex荧光基团会发出荧光(激发光535nm,发射光556nm)。
如图1所示,当cas12b与sgrna浓度为250nm或500nm时,dsdna和ssdna作为靶标都具有较高的荧光强度,当cas12b与sgrna浓度降低到50nm和100nm时,ssdna为靶标序列仍有较高荧光强度。
cas12btrans切割(反式切割)的pam特性:研究表明,cas12bcis切割双链dna时(顺式切割)需要称为pam的序列,即5'端紧接着的是5'-ttn-3'时才能被cas12b切割。为了测试cas12b在trans切割时靶标序列是否需要pam的特性,本发明人在pam位置处设计了不同序列(包括ttc,tac,atc,aac,ggc和ccc),以便检验trans切割(反式切割)时是否需要pam。
如图2所示,双链dna作为靶标时cas12b的trans切割对pam序列较为敏感,即ttc时才能有较高的trans切割荧光强度(变为tac后产生的荧光强度略低于ttc),而单链dna作为靶标时则对pam序列不敏感,始终显示出较高的trans切割荧光强度。
cas12btrans切割(反式切割)的速率:分别用ssdna和dsdna作为靶标dna时,测定cas12b的trans切割的速率。
如图3所示,可以看到,当ssdna作为底物时,cas12b的trans切割速率非常快,仅6分钟时,已接近最高值。而dsdna作为底物时,切割速度相对较慢,有一个逐渐上升的过程。这提示,对于某些需要快速检测(如≤10分钟)的场合而言,宜将ssdna作为cas12旁路切割的底物。
cas12btrans切割(反式切割)的适用温度范围:cas12b进行trans切割有非常宽的温度范围,不管是ssdna或dsdna作为靶标序列,温度从45-65℃都有很好的响应值,特别是ssdna靶标的温度范围更宽25-70℃(见图4)。
cas12b检测灵敏度:本发明人通过稀释靶标dsdna或ssdna的浓度,测试了cas12b的检测灵敏度。在一优选例中,将靶标ssdna(dnmt1-3(ttcpam)-r)或dsdna(dnmt1-3(ttcpam)-f、dnmt1-3(ttcpam)-r两寡核苷酸经退火产生)分别稀释成浓度为100nm,10nm,1nm,…到10-4nm。
结果表明,当dna浓度为1nm时,直接用cas12b进行检测,仍可以检测到靶标分子(图5)。
holmesv2.0灵敏度测试:在一实施例中,当将cas12b和lamp反应相结合(即holmesv2.0),检测的灵敏度可以达到10-8nm,大大提高了靶标核酸序列检测的灵敏度(图6)。
针对ssdna靶标的snp测试:本发明方法非常适合用于检测核酸突变,包括snp,尤其是ssdna靶标中的snp。
对于靶标单链dna的snp检测表明,当sgrna引导序列为18nt或20nt时,1-12位碱基突变后对trans切割产生的荧光信号的影响不大。当sgrna引导序列为14nt或15nt时,包括野生型对照在内的荧光值都很低。当sgrna引导序列为16nt时,10-12位突变后,其荧光值相对于对照明显下降了(图7)。
进一步研究发现,引导序列为16nt的sgrna时,10-16位碱基突变对荧光值的影响非常明显,特别是10-14位几乎检测不到荧光值(图8)。
此外,当靶标单链dna不同位置突变成不同类型的碱基,对荧光值没有本质影响,仅第9位突变成g荧光值变化较大(图9)。
另外,本发明人还测试了另外3种靶标序列,不同序列虽然对某个位点突变的荧光变化值不尽相同,但都能在第8-16位找到明显变化的位点,例如rs5082的8、10、11、12位,rs1467558的11位,rs2952768的8-15位(图10)。
针对dsdna靶标的snp测试:本发明方法适合用于检测核酸突变,包括dsdna靶标中的snp。
对于靶标双链dna的snp检测表明,单碱基突变对trans切割产生的荧光信号与ssdna不同。比如引导序列为18-20nt的sgrna,对10-16位的碱基突变有不同程度的敏感(虽然1和3位碱基突变后荧光信号也有不同程度下降)(图11)。通常,这几种sgrna对第10和16位最为敏感。
不同温度下的一步法检测:本发明还提供了将核酸扩增与基于cas12b相结合的一步法检测。
优选地,可将lamp扩增与cas12b检测结合起来,实现一步法检测。在本发明中,测试了不同温度情况下(分别为50℃,55℃,60℃,65℃),cas12b的trans切割情况。如图12所示,在50和55℃情况下,可以实现一步法检测,特别是55℃情况下,荧光值上升更快。
rna靶标的测试:本发明方法不仅适合对靶标dna的检测,还可用于对于靶标rna的检测。在本发明中,可先对rna进行反转录形成dna后,然后进行靶标序列的检测。
一种优选的方法是加入反转录酶在反应体系中,从而实现rna的反转录;然后由用例如bst2.0dna聚合酶进行扩增,并用cas12b实现trans切割。另一种方法是,直接用bst3.0dna聚合酶(此酶可以对rna模板进行扩增产生dna),从而直接实现rna的反转录和扩增。(图17)
微量dna模板的定量测试:本发明方法可用于定量检测。
在一个实例中,在55℃条件下,采用基于lamp扩增和cas12b的一步法,因此对微量dna的定量测试。通过稀释不同浓度的模板,实时检测trans切割的荧光值,通过制作标准曲线,经计算,即可得到目标模板序列的浓度(图18和19)。
另一种定量检测是将本发明的基于cas12b的检测方法与数字pcr技术进行结合(见下文“数字holmes法”)。
引物设计和sgrna设计
基于本发明的教导,本领域技术人员可以根据实际需要,合成相应的引物和/或sgrna,以便进行本发明的基于cas12b的核酸检测。在本发明中,一些优选的引物和sgrna设计可参考以下建议。
对于引物而言,其设计可参考以下:
情形1.前期扩增反应,如果选择lamp方法,则可采用常规方法进行引物设计(http://primerexplorer.jp/elamp4.0.0/index.html)。建议在引物区域f2/f3和b2/b3间增加loopf和loopb引物,即6对引物。
情形2.前期扩增反应,如果选择非对称pcr方法,则需要备选几种引物,测试最适的产生单链dna的组合。
情形3.当测试snp时,如果位点附近有合适pam位点,则用情形1的情况;如果没有合适pam位点,则可以在引物中带入pam位点,使得产物产生pam位点。通常snp位点离pam为8-16个碱基比较合适,但具体靶标序列还需具体测试。
情形4.当snp位点附近很难找到合适的lamp扩增引物或无法引入合适的pam时,可以采用非对称pcr扩增等扩增出单链dna的方法。
对于sgrna设计而言,可参考以下情况:
情形1.对于靶标基因的检测,一般宜选取20bp互补配对的sgrna,且靶标序列带pam序列。
情形2.对于snp检测,如果使用lamp等双链扩增作为扩增步骤时,宜先测试snp处于靶标序列的位置。优先测试18nt长度引导序列的sgrna,在10-16位进行测试。
情形3.对于snp检测,如果使用非对称pcr等单链扩增作为扩增步骤时,宜先测试snp处于靶标序列的位置。优先测试16nt长度引导序列的sgrna,在10-16位进行测试,特别是10-12位。
数字holmes法
在本发明中,还提供了将本发明的holmes技术与数字pcr(dpcr)相结合的技术,简称为“数字holmes法”
数字pcr是一种近年来迅速发展的核酸分子绝对定量技术。目前主要有微滴式数字pcr(ddpcr)和芯片数字pcr(cdpcr)两种,即在传统的pcr扩增前对样品进行微滴化处理或加到芯片中,将含有核酸分子的反应体系分成成千上万个纳升级的反应体系。与普通pcr相比,数字pcr多了前处理步骤和后期的荧光检测步骤。目前数字pcr检测都还需要用到普通pcr仪来进行扩增这一步,而且pcr反应的条件较为严格,且所需时间较长(大约3-4小时)。
利用holmes方法结合目前芯片数字pcr的荧光读取仪器(即数字holmes方法),只需恒温就可进行绝对定量检测。
此外,数字holmes采用lamp等方法进行扩增时,可以在一个较宽的温度范围(50-70℃,或50-55℃左右)进行扩增,而不需要像常规pcr仪那样精确的控温。由于cas12b在该温度范围内也具有旁路切割活性,因此既可以有效整合,又可同时进行扩增和旁路切割。
本发明还提供了一种用于数字holmes检测的设备,尤其是如本发明第七方面中所述的设备。
因此,利用数字holmes技术,将前处理、扩增和检测整合到一台仪器中,不仅可以减少人为操作误差,更可加快检测速度和降低仪器成本。
试剂盒
本发明还提供了一种试剂盒,包括向导rna、cas12b蛋白、核酸探针。此外,本发明试剂盒还可包括其他试剂,例如缓冲液、用于进行扩增所需的试剂、用于进行逆转录所需的试剂、或其组合。
典型地,本发明的试剂盒包括:
i)第一容器以及位于第一容器内的cas12b蛋白,所述cas12b蛋白是cas12b或者具有与cas12b的旁路单链dna切割活性类似的cas蛋白;
ii)任选的第二容器以及位于第二容器内的向导rna,所述向导rna引导所述cas蛋白特异性结合于靶标核酸分子;
iii)第三容器以及位于第三容器内的核酸探针(优选为荧光探针);
iv)任选的第四容器以及位于第四容器内的缓冲液;
其中,所述的靶标核酸分子为靶标dna。
此外,本发明的试剂盒还可包括:
v)第五容器以及位于第五容器内的用于扩增靶标dna的聚合酶;
vi)任选的第六容器以及位于第六容器内的用于反转录的反转录酶;
vii)第七容器以及位于第七容器内的用于扩增反应和/或反转录反应的dntp。
在本发明中,一个或多个或全部所述容器可以是同一或不同的容器。
本发明的主要优点在于:
利用cas12b的特性,本发明人开发了特异性检测核酸分子的方法,称之为holmesv2.0(one-hourlow-costmultipurposehighlyefficientsystemversion2.0)。其特点是快速(1小时)、低价、多通路、高效、简便。该方法可用于快速病原菌检测,snp检测等领域。相比于基于cas12a的第一代holmes,基于高温酶cas12b的第二代holmes有更多的优点。
(1)快速:在测试条件准备好的情况下,从拿到样品,到拿到检测结果只需约1小时。
(2)低成本:实验中没有特殊的材料或酶,而且涉及到的材料、试剂等较少,可以进行微量化的测试分析。
(3)高效:本发明具有极高的灵敏度,可以检测到10am浓度的dna。
(4)多用途:可检测不同的核酸样本,包括dna样本和rna样本。
(5)简单:没有特殊复杂的步骤,如果制成试剂盒以及设定好程序,只需简单的加入样品等操作。
(6)一步反应:可将扩增反应和检测反应同时反应,实现一步法测试。
(7)等温:由于lamp扩增和cas12b检测都是恒温条件,因此对于仪器要求更低。
(8)较宽的反应温度范围:cas12b的检测在45-65℃都能进行检测,因此对仪器的要求较低。
(9)两种酶即可实现dna或rna的扩增与检测:由于bst3.0dna聚合酶具有以dna或rna为模板的dna聚合酶活性,因此只需要bst3.0和cas12b两种酶即可实现dna或rna的扩增到检测,节约生产成本。
(10)高灵敏的snp检测:通过合理设计位点和sgrna,可以高灵敏的区分单个碱基的差别。
(11)可实现定量检测:通过一步法holmes进行荧光的实时检测,或结合数字pcr仪的芯片和检测系统,可实现对样本的定量检测。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
在本申请中,除非另外说明,否则cas12b反式切割活性体系为:sgrna与cas12b各250nm,反应条件为48℃30min。
材料
1.rna酶抑制剂购自takara公司,高保真dna聚合酶kodfx购自toyobo公司;引物(寡核苷酸)由上海生工合成;t7rna聚合酶购自thermo公司;rna纯化与浓缩试剂盒(rnaclean&concentratortm-5)购自zymoresearch;
2.培养基配方:液体lb(1%tryptone,0.5%yeastextract,1%nacl),配置固体lb时,只需要在液体lb中添加2%的琼脂即可。
实施例1cas12b蛋白检测双链dna(dsdna)或单链dna(ssdna)的靶标
选用单链dna或双链dna(targett1)作为靶标序列,测试不同浓度的cas12b蛋白和sgrna对其检测的响应值。
1、向导rna(sgrna)的制备
首先构建以puc18为质粒骨架构建质粒puc18-sgrna-t1,该质粒在puc18上插入了t7启动子及用于转录sgrna的模板dna序列(注:此模板转录出的sgrna靶向一个在本研究中称为t1的序列)。方法是以puc18质粒为模板,puc18-1-f和puc18-1-r为引物,先进行一轮pcr,t4dnaligase连接pcr产物,转化入dh10b中,经测序得到正确克隆,称之为puc18-sgrna-t1-pre。再以puc18-sgrna-t1-pre为模板,puc18-2-f和puc18-2-r为引物,进行第二轮pcr,同样地方法连接pcr产物并转化,最终得到测序正确的质粒puc18-sgrna-t1。
然后,以质粒puc18-sgrna-t1为模板,t7-crrna-f和sgrna-t1-r为引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna(命名为sgrna-t1),反应在37℃下过夜进行(12-16h)。
最后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、靶标dna的制备
(1)若靶标dna为单链,直接合成一条寡核苷酸targetdna(t1-r),其中包含sgrna所识别的20bp靶标序列(t1)。
(2)若靶标dna为双链,直接合成两条互补的的寡核苷酸(t1-f;t1-r),其中包含sgrna所识别的20bp靶标序列(t1)。将两条寡核苷酸退火,得到双链targetdna。具体是,将配对的寡核苷酸(2μm)在1×pcr缓冲液(transgenbiotech)中混合,总体积为20μl,然后进行退火程序:初始在95℃变性5分钟,然后从95℃冷却至20℃,使用热循环仪每分钟降低1℃。
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度,置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为50、100、250或500nm),荧光淬灭探针(hex-n12-bhq1,终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
结果如图2所示,当cas12b与sgrna浓度为250nm或500nm时,dsdna和ssdna作为靶标都具有较高的荧光强度,当cas12b与sgrna浓度降低到50nm和100nm时,ssdna为靶标序列仍有较高荧光强度。
实施例2holmesv2.0(lamp结合cas12b)测试响应灵敏度
通过对荧光探针(hex-n12-bhq1)被激发的荧光强度进行检测,确定cas12b行使旁路单链dna切割活力需要的靶标dna浓度,即cas12b旁路单链dna切割反应的灵敏度。
1、sgrna的制备
首先,以此前构建的质粒puc18-sgrna-t1为模板,设计名为sgrna-dnmt1-3-f和sgrna-dnmt1-3-r的引物,通过pcr将sgrna中靶向t1这一targetdna的20个碱基替换为靶向dnmt1-3的sgrna,得到另一质粒puc18-sgrna-dnmt1-3。
然后以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f和zlsgrna-dnmt1-3-r为引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna(命名为sgrna-dnmt1-3),反应在37℃下过夜进行(12-16h)。
最后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、靶标dna的制备
对于靶标dna,第一种是直接加入cas12b的反应体系,不经过扩增。方法如下:
(1)若靶标dna为单链,直接合成一条长50bp的寡核苷酸作targetdna(dnmt1-3(ttcpam)-r),其中包含sgrna所识别的20bp靶标序列(dnmt1-3)。
(2)若靶标dna为双链,直接合成两条互补的长50bp的寡核苷酸(dnmt1-3(ttcpam)-f;dnmt1-3(ttcpam)-r),其中包含sgrna所识别的20bp靶标序列(dnmt1-3)。将两条寡核苷酸退火,得到双链targetdna。具体是,将配对的寡核苷酸(2μm)在1×pcr缓冲液(transgenbiotech)中退火,总体积为20μl,然后进行退火程序:初始在95℃变性5分钟,然后从95℃冷却至20℃,使用热循环仪每分钟降低1℃。
(3)将单链或双链靶标dna梯度稀释为2μm,0.2μm,0.02μm,0.002μm,0.0002μm备用。
第二种是将含靶标序列(dnmt1-3)的片段插入一质粒载体中,通过lamp反应扩增。
(1)使用transgen公司的peasy-bluntzerocloningkit,将含靶标序列(dnmt1-3)片段插入peasy-bluntzerocloningvector,经测序验证后获得正确的克隆。
(2)lamp扩展反应
以上述质粒作为模板,进行lamp扩增反应,模板分别加入0,1nm,0.1nm,等10倍梯度稀释至10-11nm。每个反应体系总体积为25μl,用引物1.6μm的lamp-dnm-fip与lamp-dnm-bip、0.2μm的lamp-dnm-f3与lamp-dnm-b3、0.4μm的lamp-dnm-loopf与lamp-dnm-loopb,lamp反应所用的试剂盒为
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为250nm),1μl的targetdna或1μl的lamp产物,荧光探针(hex-n12-bhq1)(终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,置98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
如图5所示,可以看出,cas12b可以检测到1nm浓度的单链或双链dna;结合lamp扩增之后,cas12b(即holmesv2.0方法)可检测到10-8nm浓度的dna(图6)。
实施例3cas12b测试单碱基突变靶标
选用单链dna或双链dna作为靶标序列,测试当靶标dna存在单碱基突变时,cas12btrans切割荧光信号的变化,从而检测单碱基突变。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3作为模板,t7-crrna-f作为上游引物,以及含有与不同靶标互补的引导序列的寡核苷酸作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成全长sgrna(其中的引导序列长度为20nt)或截短sgrna(其中的引导序列长度小于20nt),反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、靶标dna的制备
(1)若靶标dna为单链,直接合成一条长50bp的寡核苷酸作targetdna,其中包含sgrna所识别的靶标序列。
(2)若靶标dna为双链,直接合成两条互补的长50bp的寡核苷酸,其中包含sgrna所识别的靶标序列。将两条寡核苷酸退火,得到双链targetdna。具体是,将配对的寡核苷酸(1μm)在1×pcr缓冲液(transgenbiotech)中退火,总体积为20μl,然后进行退火程序:初始在95℃变性5分钟,然后从95℃冷却至20℃,使用热循环仪每分钟降低1℃。
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为250nm),targetdna(终浓度50nm),荧光淬灭探针(hex-n12-bhq1,终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,置98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,然后用酶标仪检测(激发光535nm,发射光556nm)。
首先,就dnmt1-3这一靶标序列而言,对于靶标单链dna,如图7所示,当sgrna引导序列为20nt时,1-12位碱基突变后对trans切割产生的荧光信号的影响不大。当sgrna引导序列为14nt或15nt时,包括对照在内的荧光值都很低。当sgrna引导序列为16nt时,10-12位突变后,其荧光值明显下降了。
进一步研究发现(图8),10-16位碱基突变对荧光值的影响非常明显,特别是10-14位突变后几乎检测不到荧光值。接着,将靶标单链dna不同位置突变成不同类型的碱基,对荧光值没有本质影响,仅第9位突变成g荧光值变化较大(图9)。
接着,测试了另外3种靶标序列,不同序列虽然对某个位点突变的荧光变化值不尽相同,但都能在8-16位找到明显变化的位点,例如rs5082的8、10、11、12位,rs1467558的11位,rs2952768的8-15位(图10)。
对于靶标双链dna(如图11所示),单碱基突变对trans切割产生的荧光信号的影响与ssdna不同。以dnmt1-3这一靶标序列为例,引导序列为18-20nt的sgrna,对10-16位的碱基突变敏感。此外,在本实施例中,几种sgrna对第10和16位最为敏感。
此外,在在50℃、55℃、60℃、65℃条件下,lamp与cas12b检测实现一步法反应(图12)。
实施例4环境水体中大肠杆菌等微生物测试
选用大肠杆菌gyrb基因作为检测目标,间接测试水体中大肠杆菌等微生物,并测定检测限。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,zl-gyrb-crrna2-r作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna,反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、lamp扩增反应
将大肠杆菌mg1655培养至od600为1.0时,将一部分菌液放置100℃处理15min。然后进行lamp扩增反应,模板分别加入不同稀释程度的菌液,od600分别稀释为0,5×10-3,5×10-4,至5×10-9。
经涂板测试,5×10-3大约为7000个细菌。反应体系总体积为25μl,用引物1.6μm的lamp-gyrb-fip与lamp-gyrb-bip、0.2μm的lamp-gyrb-f3与lamp-gyrb-b3、0.4μm的lamp-gyrb-loopf与lamp-gyrb-loopb,lamp反应所用的试剂盒为
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为250nm),1μl的lamp产物,荧光探针(hex-n12-bhq1)(终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,置98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
如图13所示,结合lamp扩增之后,cas12b(即holmesv2.0方法)的灵敏度可达到检测几个大肠杆菌的程度(经过涂板测试,od600=5×10-6大约为7个大肠杆菌)。
实施例5唾液性别测试
本实施例选用了y染色体特有的sry基因位点作为测试靶标,通过holmesv2.0鉴定唾液来源样本的性别。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,zl-sry-crrna3-r和zlsgrna-dnmt1-3-r作为下游引物,通过pcr分别扩增出两种体外转录所需的dna模板。之后向pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna,反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、lamp扩增反应
分别以男性和女性的唾液作为模板,进行lamp扩增反应。每个反应体系总体积为25μl,用引物1.6μm的lamp-sry-fip与lamp-sry-bip、0.2μm的lamp-sry-f3与lamp-sry-b3、0.4μm的lamp-sry-loopf与lamp-sry-loopb,lamp反应所用的试剂盒为
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为250nm),1μl的lamp产物,荧光探针(hex-n12-bhq1)(终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,置98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
如图14所示,结合lamp扩增之后,cas12b(即holmesv2.0方法)可高效地对唾液样本进行人的性别检测。
实施例6人snp测试
在本实施例中,对人的rs5082位点进行检测。通过非对称pcr或lamp等温扩增方法制备靶标dna,鉴定唾液来源样本的snp类型。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,以及不同长度的含有与rs5082位点互补的引导序列的寡核苷酸作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成截短sgrna,反应在37℃下过夜进行(12-16h)。对于通过非对称pcr产生的靶标dna,在cas12b反应时用引导序列为16nt的截短sgrna;对于通过lamp扩增产生的靶标dna,在cas12b反应时用引导序列为18nt的截短sgrna。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、扩增反应(非对称pcr或lamp)
非对称pcr扩增反应:以人细胞基因组hek293t作为模板,用引物asp-primer,asp-rs5082-f,asp-rs5082-r进行非对称pcr扩增(而普通pcr则不加asp-primer),用kodfx酶进行扩增反应。
lamp扩增反应:以人细胞基因组hek293t作为模板,进行lamp扩增反应。每个反应体系总体积为25μl,用引物1.6μm的lamp-rs5082-fip-10pam(对照为lamp-rs5082-fip)与lamp-rs5082-bip、0.2μm的lamp-rs5082-f3与lamp-rs5082-b3、0.4μm的lamp-rs5082-loopf与lamp-rs5082-loopb,lamp反应所用的试剂盒为
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为500nm,lamp扩增测试中cas12b为250nm),1μl的targetdna或1μl的lamp产物,荧光探针(hex-n12-bhq1)(终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应60min。之后,置pcr仪中98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
如图15所示,由于rs5082位点附近没有pam序列,因此产生单链dna的非对称pcr可以更明显的区分snp位点。
如果结合lamp扩增,由于rs5082位点附近没有pam序列,需要将检测位点引入pam序列,可以更好地区分snp位点(图16)。
实施例7rna病毒测试
本实施例中,选用了rna病毒jev(japaneseencephalitisvirus;日本乙型脑炎病毒),通过holmesv2.0鉴定样本是否感染病毒,选用的位点为e453和ns170进行测试。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,以及含有与相应靶标互补的引导序列的寡核苷酸作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna,反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、lamp扩增反应
以感染jev病毒的细胞液作为模板,进行lamp扩增反应。每个反应体系总体积为25μl,用引物1.6μm的lamp-e453-fip与lamp-e453-bip、0.2μm的lamp-e453-f3与lamp-e453-b3、0.4μm的lamp-e453-loopf与lamp-e453-loopb,(或1.6μm的lamp-ns170-fip与lamp-ns170-bip、0.2μm的lamp-ns170-f3与lamp-ns170-b3、0.4μm的lamp-ns170-loopf与lamp-ns170-loopb)lamp反应所用的试剂盒为
3、cas12b反应
(1)sgrna退火:将sgrna稀释至适当浓度(5μm),置pcr仪中退火。退火程序:75℃变性5min,然后从75℃冷却至20℃,每分钟降低1℃。
(2)sgrna与cas12b孵育:将退火完成的sgrna与等摩尔浓度的cas12b混合,于30℃放置20-30min。
(3)cas12b反应:在20μl反应体系中,加入步骤(2)孵育的sgrna与cas12b的混合物(两者终浓度均为250nm),1μl的lamp产物,荧光探针(hex-n12-bhq1)(终浓度500nm),以及2μl的10×nebbuffer3.1和0.5μl的rna酶抑制剂(40u/μl)。混匀后,于48℃反应30min。之后,置pcr仪中98℃加热5min灭活。
4、荧光酶标仪法检测cas12b的旁路单链dna切割活力
将灭活的20μl反应液加入96孔板中,用酶标仪检测(激发光535nm,发射光556nm)。
如图17所示,加入lamp扩增之后,cas12b(即holmesv2.0方法)可以非常灵敏地检测是否含有jev病毒。
实施例8微量dna相对定量检测(real-timeholmes方法)
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,以及含有与靶标互补的引导序列的寡核苷酸作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna,反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、一步法lamp扩增-荧光检测反应(使用real-timepcr仪器)
以不同稀释浓度的dnmt1-3质粒作为模板,进行lamp扩增反应。每个反应体系总体积为20μl,用引物1.6μm的lamp-dnm-fip与lamp-dnm-bip、0.2μm的lamp-dnm-f3与lamp-dnm-b3、0.4μm的lamp-dnm-loopf与lamp-dnm-loopb,lamp反应所用的试剂盒为
3、结果分析
如图18所示,不同稀释浓度的靶标dna的实时荧光测定值。
以荧光值达到600,000的时间点为y轴,以浓度的lg绝对值为x轴作图,可以得到近似直线的趋势线(图19)。
上述结果表明,本发明方法可在45min内,对10-1-10-6nm的dna进行准确定量。
实施例9微量dna绝对定量检测(数字holmes方法)
在本实施例中,利用holmes方法结合芯片数字pcr,通过一个反应温度,进行绝对定量检测。
1、向导rna(sgrna)的制备
以质粒puc18-sgrna-dnmt1-3为模板,t7-crrna-f作为上游引物,以及不同长度的含有与rs5082位点互补的引导序列的寡核苷酸作为下游引物,通过pcr扩增出体外转录所需的dna模板。之后向此pcr产物加入dpnⅰ(每50μlpcr体系加入1μldpnⅰ(10u/μl)),37℃水浴30min,消化其中的质粒dna模板,并使用凝胶与pcr产物柱回收试剂盒(promega)回收pcr产物。以此pcr回收产物为模板,使用t7高产量转录试剂盒(thermo)合成sgrna,反应在37℃下过夜进行(12-16h)。
之后,在转录体系中加入dnaseⅰ(每50μl转录体系加2μldnaseⅰ(5u/μl)),37℃水浴30min,消除质粒dna模板,并使用rna纯化与浓缩试剂盒纯化rna,再用nanodrop2000c定量,于-80℃冰箱中保存备用。
2、一步法lamp扩增-荧光检测反应(使用jnmedsys的ddpcr仪器)
以不同稀释浓度的人293t细胞基因组作为模板,进行绝对定量检测。每个反应体系总体积为15μl,用引物1.6μm的lamp-rs5082-fip-10pam、lamp-rs5082-bip,0.2μm的lamp-rs5082-f3与lamp-rs5082-b3、0.4μm的lamp-rs5082-loopf与lamp-rs5082-loopb,lamp反应所用的试剂盒为
混匀后,利用滑块将样本递送至芯片上(芯片在pcr管中),进行体系分区。然后转移到sealingenhancer仪器中处理。
接着,加入235ul的封闭液,在金属浴中进行反应,条件为55℃反应120min。在0、60、90、120min时,放置到viewjig仪器中检测荧光。
3、结果分析
如图20所示,经过120分钟反应后,经计算模板1检测到有51.75个拷贝,经过10倍稀释后的模板2,经计算有6.3个拷贝,而不加模板的对照,则没有检测出拷贝数。
结果表明,采用本发明的数字holmes法,不仅可以定量检测,而且可以极其灵敏地对一个微检测体系中的单个拷贝的模板进行准确检测,且耗时大幅缩短。
表1作为底物的寡核苷酸
表2用于转录sgrna的模板序列
表3作为扩增用引物的寡核苷酸
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!