用于抗白血病的GSH/pH响应性纳米给药系统及其制备方法与流程
本发明涉及智能纳米给药系统,特别是一种用于抗白血病的gsh/ph响应性纳米给药系统及其制备方法。
背景技术:
近年来,由于具有易于优化的结构特征,聚合物前药纳米给药系统已引起了广泛关注。将疏水性药物接枝到亲水性大分子上所制备的聚合物前药,经自组装形成单组分纳米给药系统,具有下列优势:(1)提高疏水性药物的水溶性;(2)避免药物过早泄露并防止药物在血液中的降解和失活;(3)延长药物血液循环时间;(4)增强药物在靶向组织和细胞的选择性释放;(5)提高载药能力;(6)改善药物生物利用度;(7)基于epr效应增强药物在肿瘤部位的被动靶向性。
6-硫鸟嘌呤(6-tg)是一种抗代谢药物,用于急性髓细胞白血病的化疗及抗关节炎和癌症的免疫治疗。但由于其水溶性差、易代谢失活以及严重的肝毒性,6-tg的应用受到限制。在6-tg结构中含有一个伯氨基和巯基。伯氨基可与含有醛基的化合物形成席夫碱键,而巯基可与自身或其它含巯基的化合物形成二硫键。席夫碱键在中性(或弱碱性)条件下稳定存在,但在酸性条件下发生可逆断裂。由于这种特征,含有席夫碱的聚合物前药或纳米系统已经开发并用于癌症治疗,在体内细胞摄取实验中表现出ph依赖的不稳定性。然而,因为正常细胞与肿瘤细胞具有相同的酸性内环境(ph低于6.5),所以这类传递系统并不适用于癌细胞。与细胞内ph特征不同的是,肿瘤细胞中gsh的浓度比正常细胞(2-10mm)至少高出4倍,而血浆中gsh仅为微摩尔级浓度(2-20μm)。二硫键在含微摩尔水平gsh环境中稳定,但在毫摩尔级gsh浓度断开。因此,基于二硫键的给药系统可选择性将药物地送到癌细胞。
由(1-4)-β-d-甘露糖醛酸和(1-4)α-l-古洛糖醛酸组成的天然阴离子多糖海藻酸钠具有良好的水溶性、生物相容性、无毒性、相对较低的成本,已广泛用于各种生物医学应用中。据报道,与海藻酸钠形成的聚合物前药延长了药物半衰期并提高了药物溶解度。然而,由于海藻酸钠分子链具有刚性,其在体内很难降解,这使得海藻酸钠不适合作为药物载体。通过高碘酸钠氧化,制得的双醛藻酸钠不仅保留了海藻酸钠良好的水溶性、低毒性和生物相容性等优点,而且还显示出良好的生物降解性以及柔性。另外,更重要的是醛基藻酸钠可与含伯胺基的药物形成ph敏感的大分子前药。
技术实现要素:
本发明所要解决的技术问题是:为解决现有技术中存在的问题,构建一种用于抗白血病的gsh/ph响应性纳米给药系统及其制备方法。
本发明解决其技术问题采用以下的技术方案:
本发明提供的用于抗白血病的gsh/ph响应性纳米给药系统,其有效成分为疏水性抗肿瘤药6-tg与可生物降解的醛基海藻酸钠的交联化合物,6-tg是6-硫鸟嘌呤的英文缩写;
该纳米给药系统由以下方法制成:先将海藻酸钠在室温避光下经高碘酸钠氧化,制得醛基海藻酸钠,其英文缩写是dsa;然后在制得的dsa水溶液中滴加6-tg的dmf溶液,dmf是n,n-二甲基甲酰胺的英文缩写,40℃恒温反应24h,制得6-硫鸟嘌呤-醛基海藻酸钠聚合物前药,其英文缩写是6-tg-dsa;最后,将6-tg-dsa分散于去离子水中,超声处理促进巯基氧化为二硫键,获得gsh/ph双响应的二硫键交联的聚合物前药胶束,其英文缩写是ppn。
所述的醛基海藻酸钠的氧化度为20%~80%,以便提高海藻酸钠在生物体内的生物降解能力。
所述的聚合物前药胶束的粒径为118~180nm。
本发明提供的上述gsh/ph响应性纳米给药系统,用于白血病治疗的药物传递系统领域。
本发明采用以下方法制备上述的gsh/ph响应性纳米给药系统,该方法包括以下步骤:
(1)醛基海藻酸钠的制备:
将高碘酸钠与海藻酸钠糖单元按摩尔比1~5:5在去离子水中室温避光下反应24h,之后加几滴无水乙二醇使反应停止,静置2h后加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀;减压抽滤,乙醇洗涤两次,-34℃下冻干获得醛基海藻酸钠;
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备:
将醛基海藻酸钠溶于去离子水,同时将6-tg溶于dmf溶液中,按照醛基海藻酸钠的醛基与6-tg的摩尔比(1~3):1将两相混合后,滴加少量冰醋酸作为催化剂于40℃恒温水浴中加热反应24h;然后用去离子水透析3天,将产物-34℃冷冻干燥,得到6-tg-dsa;
(3)gsh/ph响应性纳米给药系统的制备:
取6-tg-dsa分散于去离子水中,然后使用超声波细胞粉碎机超声处理促进巯基氧化获得gsh/ph响应性纳米给药系统。
上述方法中,6-tg-dsa在空气的条件下超声处理,使之巯基氧化,以便提高纳米粒结构的稳定性。超声处理工艺超声30min。
上述方法中,制得的gsh/ph响应性纳米给药系统的载药率为10~35%。
上述方法步骤(1)中,是在-34℃下冻干获得醛基海藻酸钠。
上述方法步骤(2)中,是在-34℃下冷冻干燥得到6-tg-dsa。
上述方法制备的gsh/ph响应性纳米给药系统,用于白血病治疗的药物传递系统领域。
本发明基于这一转化过程,设计构建gsh/ph双响应的智能纳米给药系统,与现有技术相比具有以下的主要优点:
(1)载体材料的无毒和生物可降解性;
(2)二硫键交联能保证系统在到达靶细胞前不释放药物,使系统的形态结构被锁定,避免药物在体循环系统中提前释放,实现药物在体循环过程中的稳定性;
(3)制备聚合物前药纳米粒的过程中不使用有毒的交联剂,反应条件温和;
(4)聚合物前药纳米粒表面大量羧基在人体弱碱性环境下电离,避免粒子团聚;
(5)该系统可在细胞内弱酸性环境下,能协同响应ph和gsh刺激断裂前药分子中的亚胺键及胶束形成过程生成的二硫交联键释放6-tg。由于癌变细胞与正常细胞之间gsh浓度的差异(相差约一个数量级),可实现抗癌药物的选择性释放;
(6)该系统能提高6-巯鸟嘌呤的水溶性。
(7)该系统的核壳结构还可以负载其他抗癌药物,实现双药控释,降低单药的耐药药性。
附图说明
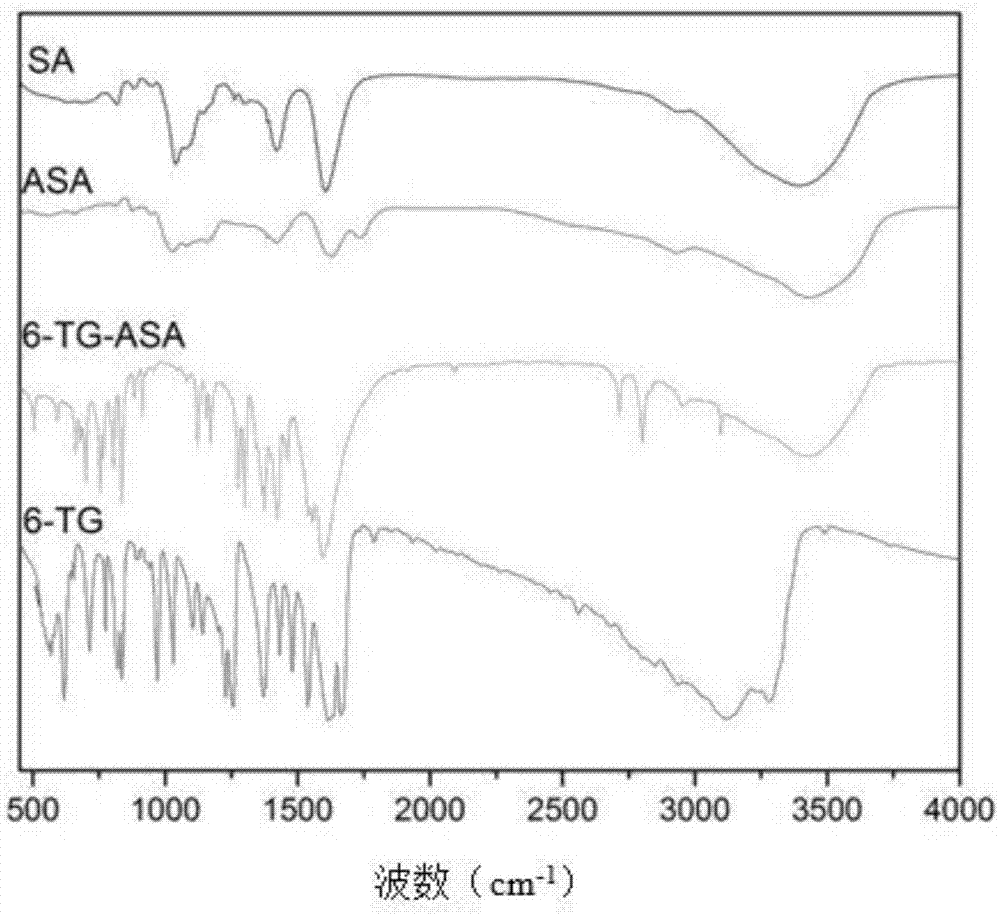
图1为本发明制备的6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的红外吸收光谱图。
图2为本发明制备的6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的核磁共振氢谱图。
图3为本发明制备的gsh/ph双响应智能纳米给药系统的透射电镜图。图中,(a)ph7.4;(b)ph5.0。
图4为本发明制备的gsh/ph双响应智能纳米给药系统ppn-3在不同ph和gsh浓度环境中随培育时间的6-tg释放变化曲线图。图中,(■):ph7.4和0.0mmgsh;(●):ph7.4和0.01mmgsh;(▲):ph=5.0和0.0mmgsh;(▼):ph=5.0和10mmgsh;(○):ph=5.0和20mmgsh。
图5为本发明制备的gsh/ph双响应智能纳米给药系统ppn-1、ppn-2、ppn-3在含有20mmgsh且ph5.0环境中随培育时间的6-tg释放变化曲线图。图中,(■):ppn-1;●):ppn-2;(▲):ppn-3。
图6为本发明制备的空白dsa对hl-60细胞(a)和l-o2细胞(b)的细胞毒性,将细胞与dsa以100至900ug/ml的浓度培育48小时。将6-tg和ppm对(c)hl-60细胞和(d)l-o2细胞的细胞毒性,将细胞与不同浓度(10-100μg/ml)的聚合物前药胶束一起培育48小时,以游离6-tg用作对照。误差栏代表标准偏差(n=6)。
具体实施方式
本发明基于6-硫鸟嘌呤和醛基藻酸钠构建了用于白血病治疗的新型ph/gsh响应性聚合物前体药物纳米系统。首先通过高碘酸钠氧化sa合成dsa,然后与疏水药物6-tg通过席夫碱反应形成两亲性聚合物前药。该前药在去离子水中自组装形成核壳纳米颗粒,并经超声氧化巯基交联形成二硫键交联纳米粒。
本发明所要解决的技术问题是:(1)6-巯鸟嘌呤是一种疏水性抗癌药物,在体内生物利用度低;(2)6-巯鸟嘌呤对肿瘤细胞的选择性差,在治疗白血病过程中有较严重的毒副作用。
下面结合实施例及附图对本发明作进一步说明,但不限定本发明。
实施例1:
(1)醛基海藻酸钠(dsa)的制备:
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠1.0796g,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为80%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备:
称取制得的醛基海藻酸钠0.01g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物于-34℃冷冻干燥,即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为35.0%。
(3)gsh/ph响应性纳米给药系统的制备:
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,命名为ppn-1。胶束的粒径大小为118nm。
实施例2:
(1)醛基海藻酸钠(dsa)的制备
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠1.0796,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为80%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备
称取制得的醛基海藻酸钠0.02g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物于-34℃冷冻干燥。即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为29.3%。。
(3)gsh/ph响应性纳米给药系统的制备
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,命名为ppn-2。胶束的粒径大小为127nm。
实施例3:
(1)醛基海藻酸钠(dsa)的制备
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠0.499g,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为60%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备
称取制得的醛基海藻酸钠0.01g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物于-34℃冷冻干燥。即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为21.2%。
(3)gsh/ph响应性纳米给药系统的制备
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,ppn-3。胶束的粒径大小为139nm。
实施例4:
(1)醛基海藻酸钠(dsa)的制备
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠0.499g,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为60%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备
称取制得的醛基海藻酸钠0.03g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物冷冻干燥。即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为15.7%。
(3)gsh/ph响应性纳米给药系统的制备
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,命名为ppn-4。胶束的粒径大小为153nm。
实施例5:
(1)醛基海藻酸钠(dsa)的制备
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠0.214g,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为20%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备
称取制得的醛基海藻酸钠0.01g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物于-34℃冷冻干燥。即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为14.9%。
(3)gsh/ph响应性纳米给药系统的制备
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,命名为ppn-5。胶束的粒径大小为164nm。
实施例6:
(1)醛基海藻酸钠(dsa)的制备
取1.00g海藻酸钠(sa)加入到20ml去离子水中,加热搅拌使其溶解。冷却到室温,使用容量瓶定容到100ml。称取高碘酸钠0.214g,加入锥形瓶中和海藻酸钠溶液在避光磁力搅拌条件下反应24h。之后加几滴无水乙二醇使反应停止,静置2h后,再加入无水乙醇和少许nacl固体,搅拌后静置至出现絮状沉淀。减压抽滤,并用少许无水乙醇洗涤两次,于-34℃冷冻干燥获得醛基海藻酸钠。氧化度为20%。
(2)6-巯鸟嘌呤-醛基海藻酸钠聚合物前药的制备
称取制得的醛基海藻酸钠0.03g溶于2ml去离子水,同时称取0.034g6-巯鸟嘌呤(6-tg)溶于8mldmf中,两相混合后滴加32μl无水冰醋酸作为催化剂于40℃恒温水浴中加热反应24h。使用透析袋透析三天后,将产物于-34℃冷冻干燥。即得到6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药。载药率为10%。
(3)gsh/ph响应性纳米给药系统的制备
将5.0mg的6-tg-dsa分散于5.0ml的缓冲溶液(pbs,10mm,ph7.4)中。将所得溶液装入10ml塑料ep管中,然后使用超声波细胞粉碎机超声处理促进巯基的氧化反应,获得聚合物前药胶束,命名为ppn-6。胶束的粒径大小为180nm。
1、醛基海藻酸钠(dsa)氧化度的测定:
取实施例1至实施例6制备的醛基海藻酸钠约0.1g,将其加入25ml的0.25mol/l的盐酸羟胺-甲基橙溶液中,充分搅拌使其溶解成均一溶液,静置24h后,以0.1mol/l标准naoh水溶液为滴定剂,采用电位滴定法测定溶液中所释放hcl的量,当溶液从红色变成黄色后停止滴定。以naoh溶液的体积v为横坐标、ph值为纵坐标,绘制ph-v滴定曲线,再对该曲线进行一次微分,所得微分曲线的峰值即为滴定终点时所消耗的naoh溶液体积v。醛基浓度(c-cho,mol/g)可通过下式(1)、(2)计算:
其中,δv为滴定时所消耗的naoh溶液的体积,单位l;nnaoh为naoh溶液的摩尔浓度,单位mol/l;n-cho为海藻酸钠上醛基的摩尔数;m海藻酸钠为海藻酸钠的质量,单位g。由醛基浓度可计算出氧化程度(oxidationdegree,od),即被氧化的海藻酸钠单体单元与总的单体单元摩尔数之比。
2、gsh/ph双响应的海藻酸钠聚合物前药胶束在不同内环境的药物释放研究:
将实施例1至实施例6制备的每个样品分成五份样品环境中,使其终浓度为1.0mg/ml,然后立即转入透析袋中。将透析袋浸入50ml相应的离心管中。实验在37±0.1℃的恒温水浴和50rpm的振荡速度下进行。在整个释放实验期间,定期取样10μl,并补充相应的空白溶液。对于不含gsh和10μmgsh的样品的离心管,首先用含有10mm半胱氨酸的相应ph溶液将10μl样品稀释至5ml,然后在振荡浴中再孵育4小时以完全将释放的tgsstg(6-tg的二硫化物二聚体)转化为6-硫鸟嘌呤(6-tg),然后再取样采用高效液相色谱法分析。
药物的释放的环境分别为:不含gsh的ph7.4和5.0缓冲溶液(用作对照);ph7.4且含10mmgsh(人血液环境)的缓冲溶液;ph5.0含有10mmgsh(正常细胞的细胞内环境)和ph5.0含有20mmgsh(癌细胞的细胞内环境)的缓冲溶液。
6-tg的高效液相色谱条件:所用仪器为高效液相色谱仪(agilent1100),紫外检测波长为248nm。色谱柱为柱altimac18柱(4.6mm×250mm,5μm);,流动相为0.05mol·l-1磷酸二氢钠溶液(磷酸调ph3.0);柱温为35℃;流速1.0ml·min-1。
如下公式计算累积药物释放:
其中mt是在时间t释放的药物量;m0是胶束中药物的初始量。释放实验进行三组平行实验,所呈现的结果是平均数据。
3、gsh/ph双响应的海藻酸钠聚合物前药胶束的体外细胞毒性研究:
通过mtt分析评估聚合物前药胶束对hl-60细胞的细胞毒性。将hl-60细胞接种于100μl含有10wt%胎牛血清的dmem培养基中的96孔板(每孔1×104个细胞)中,并在37℃,5%co2条件下培养24小时。之后,将培养基替换为200μl含有浓度范围100-900ug/ml的dsa,药物浓度范围10-100ug/ml的聚合物前药胶束和游离6-tg的新鲜培养基等效的剂量范围。将细胞再培养48小时后进行mtt测定。在酶标仪(model680)上于490nm处测量溶液的吸光度。通过比较490nm处的吸光度和仅含有细胞培养基的对照孔来确定相对细胞活力。数据表示为平均值±sd(n=6)。
细胞存活率按下列公式计算:
式中,v为细胞存活率;odt为dsa、6-tg以及聚合物前药胶束条件下的吸光值;odc为对照样品的吸光值。
图1显示了海藻酸钠(sa)、醛基海藻酸钠(dsa)、gsh/ph双响应的聚合物前药6-tg-dsa和6-硫鸟嘌呤(6-tg)的红外谱图。曲线dsa和曲线sa对比发现在1735cm-1处出现新的吸收峰,这是是醛基的特征吸收,表明形成了醛基海藻酸钠。与曲线dsa相比,曲线6-tg-dsa在500和1600cm-1之间表现出许多尖锐和密集的吸收带,这与曲线6-tg高度相似。同时曲线6-tg-dsa和曲线6-tg相比,在3000-3400cm-1间代表氨基的双峰消失,这说明6-tg的氨基参与反应;此外,曲线6-tg-dsa中1735cm-1处醛基的特征峰消失,表明醛基也参与反应,也就是说6-tg键合到dsa分子链上。
图2显示海藻酸钠(sa)(重水为溶剂)、6-硫鸟嘌呤(6-tg)(氘代dmso为溶剂)、醛基海藻酸钠(dsa)(重水为溶剂)、6-硫鸟嘌呤-醛基海藻酸钠(6-tg-dsa)聚合物前药(重水:氘代dmso=1:10的比例混合而成的混合试剂作溶剂)的核磁氢谱图。与sa的谱图相比,dsa谱图中形成的新的信号峰δ=8.3ppn,归属于醛基的质子,而在δ=5.15ppm和δ=5.4ppm处出现的新信号峰则对应于醛和邻近羟基形成的半缩醛质子,证实了sa被氧化形成dsa。在6-tg-dsa谱图中,在δ=7.9ppm处的信号峰归属于6-tg环上的次甲基质子,表明了6-tg接枝到dsa链上,此外,代表6-tg环上的氨基质子的特征信号δ=6.4ppm(参见6-tg光谱)和δ=8.3ppm处代表醛基质子的信号峰消失,表明在6-tg和dsa之间形成席夫碱键,从而生成了新的化合物6-tg-dsa。
图3显示了聚合物前药胶束在不同ph下的透射电镜图。在ph7.4条件下的胶束具有球形结构和相对均匀的尺寸,并且它们的平均尺寸约为98.4±4.87nm(参见图3a)。在ph5.0条件下的样品显示出相较而言更小的且具有更粗糙壳表面的核(参见图3b),表明胶束核的部分因酸性条件水解。
图4显示了聚合物前药胶束在五种溶液中样品的药物释放曲线。在模拟正常人血环境的条件和下,仅观察到极少量6-tg从聚合物前药胶束中释放出来;在模拟正常细胞内环境和癌细胞的细胞内环境的两组中,均能明显观察到6-tg的释放;在模拟癌细胞内环境的组中,6-tg在一定时间内的释放速率和释放药量明显高于其他所有组。
图5显示了在含有20mmgsh和ph5.0缓冲溶液中具有不同6-tg含量的聚合物前药胶束样品的累积释放药物曲线。结果显示,来自三个样品的6-tg的累积释放速率显示出显着的差异,按照以下顺序递减:ppn-3>ppn-2>ppn-1。
图6中:(a)、(b)显示了空白dsa载体(100-900μg/ml)在孵育48小时后对l-o2正常细胞和hl-60癌细胞均没有显示出明显的细胞毒性。这表明dsa在100-900μg/ml浓度范围内无毒性或有轻微毒性。然而,对于低浓度(100μg/ml)的聚合物前药胶束,我们能观察到其对hl-60癌细胞略高于具有等效剂量游离6-tg的细胞抑制率(c),表明聚合物胶束前药具有较高的细胞毒性。这样的实验结果可能归因于癌细胞中高gsh浓度和酸性环境,它们共同导致了聚合物前药胶束的解体以及共价键缀合的6-tg的释放。此外,聚合物前药胶束更强的渗透性也促进了6-tg的细胞毒性。与hl-60细胞的结果相反,用游离的6-tg和聚合物前药胶束孵育l-o2细胞48小时后,前者的细胞毒性明显高于后者(b),这表明聚合物前药胶束可显着抑制6-tg的细胞毒性并保护l-o2细胞,该结果显然归因于l-o2细胞中较低的gsh浓度导致较低浓度的6-tg释放,因此观察到较低的细胞毒性。
- 还没有人留言评论。精彩留言会获得点赞!