一种氧化还原一体化制备喹啉化合物的方法与流程
本发明属于有机合成技术领域,具体涉及到一种氧化还原一体化制备喹啉化合物的方法。
背景技术:
喹啉类化合物及其衍生物是一类重要的有机化合物,其被广泛地用于功能药物、农药、染料、化学试剂、光学材料和功能聚合物等的合成(andriesketal.science.2005;307(5707):223-27;theeraladanoncetal.tetrahedron:asymmetry.2005;16(4):827-31;romagetal.europeanjournalofmedicinalchemistry.2000;35(11):1021-35;zhangxjetal.macromolecules.1999;32(22):7422-29)。其合成方法一直受到了人们的广泛关注,其中不乏有机多个有机人名反应skraup反应(organicreactions.1953;7:59-98),doebner反应(j.chem.soc.1934:1520-23),doebner-vonmiller反应(berichtederdeutschenchemischengesellschaft.1883;6(2):2464-72),combes反应(j.chem.soc.1927:1832-57),conrad-limpach反应(chem.rev.1942;30(1):113-44),
然而,这种直接合成喹啉的策略后来被证实是相当困难的,主要的原因在于串联反应的复杂性。目前为止,只有数个使用均相ru(chem.soc.jpn.1984,57,435-438)、rh(organometallics1982,1,1003-1006)化合物和多相ir/tio2(angew.chem.int.ed.2011,50,10216-10220)和pt-sn/al2o3催化体系(chin.j.catal.2013,33,1423-1426)。以及tio2、酸化-tio2、n-dopingtio2和tio2担载贵金属的光催化体系(arabjchem2017,10,s28-s34;acscatal.2013,3,565-572;rscadv.2012,2,2848-2855;catalcommun2011,12,389-393;chem.soc.jpn.2011,84,953-959)可以实现以上的过程。通过分析,一条比较经济可行的方案是使用一种廉价易得且稳定的非贵金属多相催化剂来实现直接转化的过程。
本发明涉及一种氧化还原一体化制备喹啉化合物的方法。该方法以芳香硝基化合物和脂肪醇为原料,以含氧二硫化钼为催化剂,在惰性气氛或者在含氧的气氛,150~200℃条件下反应2~12h,反应完成后,分离液相成分,浓缩,经硅胶柱分离,得到取代喹啉化合物。该合成方法在喹啉化合物合成方面可能有重要的应用。
技术实现要素:
本发明涉及一种氧化还原一体化制备喹啉化合物的方法。该方法以芳香硝基化合物和脂肪醇为原料,以含氧二硫化钼为催化剂,在惰性气氛或者在含氧的气氛,150~200℃条件下反应2~12h,反应完成后,分离液相成分,浓缩,经硅胶柱分离,得到取代喹啉化合物。该合成方法在喹啉化合物合成方面可能有重要的应用。
针对芳香硝基化合物,其可以为:(1)硝基苯及硝基苯取代物rx-(c6h5-x)-no2(x=1~5),其中,r代表不同的取代基(r=h,f,cl,br,i,ch3,och3,nh2,no2,cho,ph等),x代表取代基的数目。当x>1是r可以代表的相同的取代基也可以代表不同的取代基;(2)苯环被其它芳香稠环取代的硝基化合物,其它芳香稠环可以为萘环,蒽环等中的一种或者多种;(3)苯环被芳香杂环取代的硝基化合物,可以为吡啶环,噻吩环,呋喃环,咪唑环等中的一种或者多种。
针对脂肪醇,其在该过程中同时作为溶剂和反应物。脂肪醇为h(ch2)nch2ch3oh(0≤n≤6)中一种或二种以上。当n>2是,除了直链烷烃取代基外,还可以是带有支链的取代基。
针对催化剂为含有氧二硫化钼的催化剂为mos2-xoy,通过控制钼酸盐前驱物的硫化与还原程度,所得催化剂中0≤x≤0.4和0≤y≤0.2。关于硫化钼催化剂的制备方法:以钼酸铵和/或钼酸钠为前驱物,硫脲和/或硫化钠为硫源,按照n(mo):n(s)=1:3~1:30的摩尔比例分散于水溶液中,在160~240℃下水热处理6~72h,过滤洗涤得到催化剂。其中以钼酸铵为前驱物效果较钼酸钠效果较好,硫源中硫脲价格便宜且容易控制分解,在制备硫化钼材料过程中更为合适,对催化剂剂的合成进行优化:按照n(mo):n(s)=1:6~1:30的摩尔比投料,在160~220℃下水热处理12~48h较为合适。
针对具体的合成条件,脂肪醇同时为底物和反应溶剂,芳香硝基化合物底物的浓度为0.001~5mol/l,催化剂的用量为芳香硝基化合物底物质量的0.5~20w%,反应温度为140℃~220℃,反应时间为1h~12h。其较为优化的条件为,芳香硝基化合物底物的浓度为0.05~2mol/l,催化剂的用量为芳香硝基化合物底物质量的0.5~20w%,反应温度为140℃~200℃,反应时间为2h~12h。
针对具体的合成条件,反应气氛可以为纯惰性气氛,n2、ar或者两者的混合气,压力在0.1~3mpa;或者反应在含氧的气氛下进行,空气或者o2气氛,压力在0.1~1mpa。
本发明属于有机合成技术领域,具体涉及到一种氧化还原一体化制备喹啉化合物的方法。
附图说明
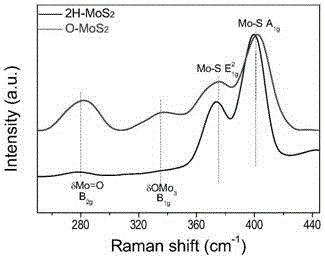
附图1:商业mos2(黑色2h-mos2)和mos2-xoy(180-24h)(红色o-mos2)的raman图谱。对比显示mos2-xoy(180-24h)中有明显的晶格氧的残留或者掺入。
附图2:mos2-xoy(180-24h)的edx元素分析,分析后的催化剂表示形式为mos1.73o0.1(180-24h)。
具体实施方式
为了对本发明进行进一步详细说明,下面给出几个具体实施案例,但本发明不限于这些实施例。
首先,为了方便表述使用的催化剂,对相关催化剂进行描述。mosx基催化剂是通过水热的方式进行合成的。商业化钼酸铵以及硫脲按照n(mo):n(s)=1:3~1:30加入到一个150ml四氟内衬的不锈钢高压釜中,在搅拌的条件下加入90ml去离子水。然后密闭后不锈钢高压釜被放置在160~250℃的烘箱内处理12~48h。处理完成后,反应釜自然冷却至室温,黑色固体用去离子水与无水乙醇洗涤。得到的催化剂命名为mos2-xoy(m-nh),其中m代表处理的温度,n代表处理的时间。为了验证催化剂中是否含有晶格氧的残留或者掺入,首先使用raman光谱对所得到的催化剂进行表征,随后使用edx电镜表征技术对催化剂中的含氧量进行表征。mos2-xoy(m-nh),相关结果标注于催化剂的x参数和y参数。以mos2-xoy(180-24h)的表征为例,附图1为其raman光谱图,附图2为其元素分布图。
附图1:商业mos2(黑色2h-mos2)和mos2-xoy(180-24h)(红色o-mos2)的raman图谱。对比显示mos2-xoy(180-24h)中有明显的晶格氧的残留或者掺入。
附图2:mos2-xoy(180-24h)的edx元素分析,分析后的催化剂表示形式为mos1.73o0.1(180-24h)
针对反应的实施例:
反应方程式:
实施例1:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入1atm的ar气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为90%,烷基取代喹啉(2-甲基喹啉)的分离收率为50%。
实施例2:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入1atm的air气,190℃下,反应6小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为96%,烷基取代喹啉的分离收率为71%。
实施例3:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为>99%,烷基取代喹啉的分离收率为72%。
实施例4:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的ar气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为80%,烷基取代喹啉的分离收率为45%。
实施例5:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.85o0.02(200-24h),置于25ml反应釜中,置换气体,充入1atm的ar气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为60%,烷基取代喹啉的分离收率为39%。
实施例6:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.85o0.02(200-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为90%,烷基取代喹啉的分离收率为63%。
实施例7:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.99o0(200-48h),置于25ml反应釜中,置换气体,充入1atm的ar气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为60%,烷基取代喹啉的分离收率为20%。
实施例8:
将0.5mmol硝基苯、2.5ml乙醇以及25mg催化剂mos1.95o0(200-48h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为72%,烷基取代喹啉的分离收率为40%。
实施例9:
将0.5mmol对甲基硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入1atm的ar气,190℃下,反应8小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。对甲基硝基苯的转化率为92%,烷基取代喹啉的分离收率为30%。
实施例10:
将0.5mmol对甲基硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。对甲基硝基苯的转化率为>99%,烷基取代喹啉的分离收率为25%。
实施例11:
将0.5mmol对氯硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。对氯硝基苯的转化率为>99%,烷基取代喹啉的分离收率为55%。
实施例12:
将0.5mmol对氯硝基苯、2.5ml乙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的o2气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。对氯硝基苯的转化率为>99%,烷基取代喹啉的分离收率为45%。
实施例13:
将0.5mmol硝基苯、2.5ml正丙醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为>99%,烷基取代喹啉的分离收率为73%。
实施例14:
将0.5mmol硝基苯、2.5ml正丁醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为>99%,烷基取代喹啉的分离收率为64%。
实施例15:
将0.5mmol硝基苯、2.5ml正戊醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为>99%,烷基取代喹啉的分离收率为55%。
实施例16:
将0.5mmol硝基苯、2.5ml正己醇以及25mg催化剂mos1.73o0.1(180-24h),置于25ml反应釜中,置换气体,充入4atm的air气,190℃下,反应4小时,离心,使用gc对液相样品进行分析,随后将液相成分浓缩,使用硅胶柱对产物进行分离。硝基苯的转化率为>99%,烷基取代喹啉的分离收率为58%。
- 还没有人留言评论。精彩留言会获得点赞!