3,4-二氢-1-吡咯啉-N-氧化物衍生物合成方法与流程
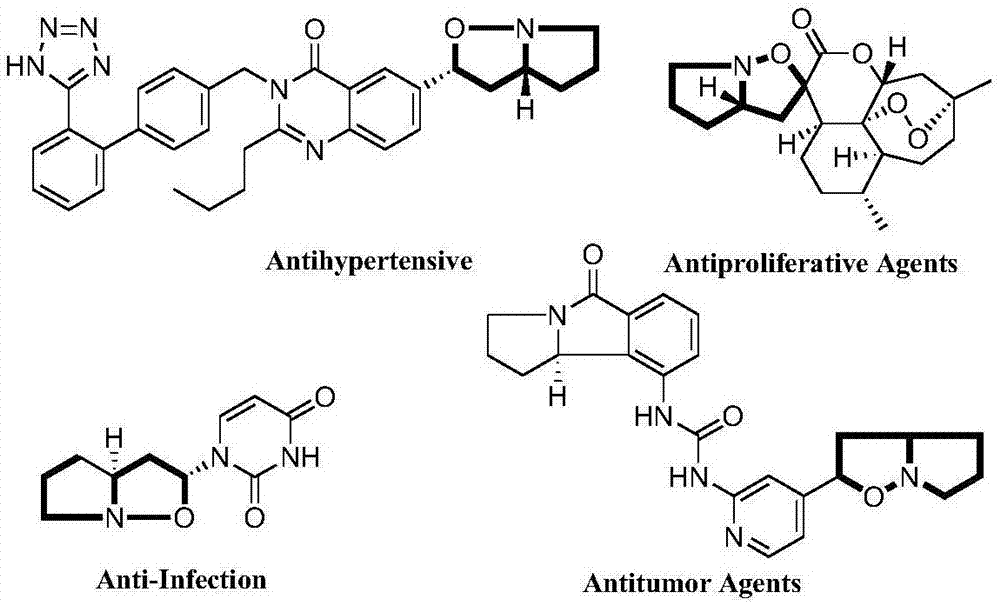
本发明属于有机中间体合成领域,具体涉及一种3,4-二氢-1-吡咯啉-n-氧化物衍生物的合成方法。
背景技术
3,4-二氢-1-吡咯啉-n-氧化物作为一种重要的1,3-偶极化合物在药物化学及天然产物合成中占有非常重要的地位,是构筑吡咯并[1,2-b]异恶唑结构单元的关键反应前体(附图1)。因此,这类化合物的合成受到广泛的关注。
目前,3,4-二氢-1-吡咯啉-n-氧化物的合成方法主要有两种(附图2),一种是通过环状仲胺、羟胺的氧化来制备3,4-二氢-1-吡咯啉-n-氧化物,反应中要用到m-cpba,oxone等强氧化剂(org.lett.,2015,17,4964–4967)。该方法反应条件剧烈,官能团耐受性差,产率及区域选择性也不好;另一种方法是通过羟胺与羰基化合物的分子内缩合反应来合成3,4-二氢-1-吡咯啉-n-氧化物,该方法条件相对较温和,但从产物结构多样性角度而言,此方法显得较为复杂(tetrahedron,2010,66,3608-3613)。
技术实现要素:
本发明要解决的技术问题是克服现有方法的缺陷,提供了一种3,4-二氢-1-吡咯啉-n-氧化物衍生物的合成方法。该方法在合成多取代五元3,4-二氢-1-吡咯啉-n-氧化物中具有官能团耐受性好、底物适用性强、合成效率高的优点。
为了解决上述技术问题,本发明提供了如下的技术方案:
一种3,4-二氢-1-吡咯啉-n-氧化物衍生物的合成方法,包括:在路易斯酸的促进下,由式i所示的取代二恶唑与式ii所示的取代环丙烷反应得到式iii所示的3,4-二氢-1-吡咯啉-n-氧化物衍生物,反应式如下
其中,
r1为氢、烷基、烷氧基、呋喃基、噻吩基或芳基;
r2为芳基、烷基或烯基;
r3和r4为各自独立的吸电子基团。
优选地,所述路易斯酸为三氯化铝、四氯化钛、三氯化镓、三氯化铟、三氯化铁、三氟甲磺酸锌、三氟甲磺酸镱、三氟甲磺酸铜、三氟甲磺酸钪、三氟甲磺酸铟、三氟甲磺酸钇、三氟甲磺酸铪、三氟甲磺酸镧、三氯化铁、三氟化硼乙醚、高氯酸镍、氯化锌。。
优选地,所述吸电子基团为酯基、酰基或氰基、磺酰基、亚磺酰基、硝基。
更优选地,所述酯基为甲酯基、乙酯基、异丙酯基、苯酚酯基、叔丁酯基、苄酯基、2,2,2-三氟乙醇酯基、二苯甲醇酯基,所述酰基为甲酰基、乙酰基、丙酰基、苯甲酰基,所述磺酰基与亚磺酰基结构式分别如下:
优选地,所述烷基为c1-c10的烷基,所述烷氧基为c1-c10的烷氧基,所述烯基为c2-c10的烯基,所述的芳基为苯或取代苯基。
所述的取代苯基如烷基取代苯基、烷氧基取代苯基、卤代苯基等,例如4-甲基苯基、4-乙基苯基、4-甲氧基苯基、4-乙氧基苯基、对氟苯基、对氯苯基。
优选地,所述反应以非质子性溶剂为反应介质。
优选地,所述路易斯酸与式ii所示的取代环丙烷的摩尔比为0.01:1~5:1,优选为0.05:1~0.2:1。
优选地,在反应体系中加入用于脱水的分子筛。
附图说明
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
附图1是含有吡咯并[1,2-b]异恶唑结构单元的活性化合物。
附图2是3,4-二氢-1-吡咯啉-n-氧化物的常规合成方法。
附图3是实施例1所述3,4-二氢-1-吡咯啉-n-氧化物3aa的x-射线单晶衍射结构;
附图4是实施例1所述3,4-二氢-1-吡咯啉-n-氧化物3aa的核磁共振氢谱数据。
附图5是实施例1所述3,4-二氢-1-吡咯啉-n-氧化物3aa的核磁共振碳谱数据。
附图6是实施例2所述3,4-二氢-1-吡咯啉-n-氧化物3ed的核磁共振氢谱数据。
附图7是实施例2所述3,4-二氢-1-吡咯啉-n-氧化物3ed的核磁共振碳谱数据。
附图8是实施例3所述3,4-二氢-1-吡咯啉-n-氧化物3gc的核磁共振氢谱数据。
附图9是实施例3所述3,4-二氢-1-吡咯啉-n-氧化物3gc的核磁共振碳谱数据。
具体实施方式
以下结合附图对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
实施例1
在50ml圆底烧瓶中加入1.0g充分干燥的
1hnmr(400mhz,cdcl3)δ8.30–8.27(m,2h),7.42–7.36(m,8h),5.38(t,j=8.0hz,1h),3.81(s,3h),3.71(s,3h),3.37(dd,j=13.2hz,8.0hz,1h),2.83(dd,j=13.6hz,8.0hz,1h);13cnmr(100mhz,cdcl3)δ169.4,168.9,138.8,136.6,130.2,128.9,128.2,128.0,127.9,76.4,76.3,63.3,53.7,53.5,53.4,38.3,38.2;hrmscalcdforc20h20no5[m+h]+:354.1336,foundfor:354.1338.
实施例2
在10ml圆底烧瓶中加入200mg充分干燥的
1hnmr(400mhz,cdcl3)δ7.53(dd,j=6.8hz,2.0hz,2h),7.32–7.16(m,7h),5.16(t,j=8.4hz,1h),3.79(s,3h),3.78(s,3h),3.25(dd,j=14.0hz,8.8hz,1h),3.05–2.84(m,4h),2.63(dd,j=14.0hz,8.0hz,1h);13cnmr(100mhz,cdcl3)δ168.5,168.0,142.7,141.0,135.6,132.1,129.3,128.4,126.1,123.1,74.2,63.1,53.6,53.5,35.3,29.5,29.4;hrmscalcdforc22h23brno5[m+h]+:460.0754,foundfor:460.0760.
实施例3
在10ml圆底烧瓶中加入200mg充分干燥的
1hnmr(400mhz,cdcl3)δ7.55(d,j=4.0hz,1h),7.52(d,j=4.8hz,1h),7.38(d,j=8.4hz,2h),7.28–7.25(m,2h),7.17(t,j=4.8hz,1h),5.37(t,j=8.0hz,1h),3.82(s,3h),3.80(s,3h),3.50(dd,j=13.6hz,8.4hz,1h),2.77(dd,j=13.2hz,8.0hz,1h);13cnmr(100mhz,cdcl3)δ169.0,168.0,135.1,134.8,129.3,129.2,129.1,128.9,126.5,73.6,62.5,53.7,53.6,38.0;hrmscalcdforc18h17clno5s[m+h]+:394.0510,foundfor:394.0513.
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!