依洛昔巴特的结晶修饰物的制作方法
本发明涉及关于n-{(2r)-2-[({[3,3-二丁基-7-(甲硫基)-1,1-二氧化-5-苯基-2,3,4,5-四氢-1,5-苯并噻庚因-8-基]氧基}乙酰基)氨基]-2-苯基乙醇基}甘胺酸(依洛昔巴特(elobixibat))的结晶修饰物、更具体而言结晶修饰物i、iv、meoh-1、etoh-1、1-proh-1及2-proh-1。本发明还涉及关于制备这些结晶修饰物的方法及包含结晶修饰物iv的药物组合物。
背景技术
wo02/50051公开了化合物1,1-二氧基-3,3-二丁基-5-苯基-7-甲硫基-8-(n-{(r)-1'-苯基-1'-[n'-(羧甲基)氨基甲酰基]甲基}氨基甲酰基甲氧基)-2,3,4,5-四氢-1,5-苯并噻庚因(依洛昔巴特;iupac名称:n-{(2r)-2-[({[3,3-二丁基-7-(甲硫基)-1,1-二氧化-5-苯基-2,3,4,5-四氢-1,5-苯并噻庚因-8-基]氧基}乙酰基)氨基]-2-苯基乙醇基}甘胺酸)。此化合物涉及回肠胆汁酸转运体(ibat)抑制剂,其可用于治疗或预防诸如异常血脂症、便秘、糖尿病及肝病等疾病。根据wo02/50051的实验部分,制备依洛昔巴特中的最后合成步骤涉及由在酸性条件下水解叔丁氧基酯组成。藉由在减压下蒸发反应混合物并藉由制备型hplc使用乙腈/乙酸铵缓冲液(50:50)作为洗脱剂纯化残余物来获得粗制化合物(实例43)。在冷冻干燥产物后,未鉴别出结晶物质。
期望发现在稳定性方面足够适于配制为药物的依洛昔巴特形式。
在形成本发明的基础的结晶研究中,使用x射线粉末衍射(xrpd)技术观察到依洛昔巴特藉由在结构中纳入溶剂分子可自许多溶剂或溶剂混合物结晶,藉此形成特定溶剂合物或混合型溶剂合物。因此,在许多溶剂或溶剂组合中获得依洛昔巴特的不同结晶修饰物。当使用相同溶剂时甚至还可获得不同结晶修饰物。此外,使用热重分析(tga)可推断出,相同结晶修饰物的不同试样可含有不同量的溶剂。当自结晶溶剂合物蒸发所纳入的有机溶剂分子时可获得依洛昔巴特的其它结晶修饰物。因此,支持本申请案的实验工作发现,依洛昔巴特的许多结晶修饰物不稳定,及/或观察到其转化为其它结晶修饰物。因此,藉由重复类似实验难以获得一致结果。
可藉由以下揭露不同的溶剂化结晶修饰物:使用极快x射线检测器并自欲至试样架上分析的固体材料浆液提取湿试样,将试样保持在实验温度下且然后在试样干燥时迅速且重复分析该试样。此技术可显示初始形成的溶剂合物或混合型溶剂合物、去溶剂化修饰物或两者的混合物。若存在一种以上部分或完全去溶剂化的结晶修饰物,则xrpd数据甚至更可能发生变化。因此,仅获得纯结晶修饰物的xrpd数据涉及另一挑战。
各种结晶修饰物可能具有众多缺点,包括结晶度易变及处置与配制困难。因此,业内需要稳定性、散装处置及溶解性方面的性质有所改良的稳定的依洛昔巴特结晶修饰物。因此,本发明的目标涉及提供稳定且高度结晶的依洛昔巴特结晶修饰物。
技术实现要素:
本发明提供依洛昔巴特(elobixibat)的各种结晶修饰物。在一方面中,结晶修饰物涉及依洛昔巴特的单水合物。单水合物包括与每摩尔依洛昔巴特结晶结合的0.9-1.1摩尔水。此处所计算水的量不包括吸附至晶体的表面的水。在某些实施方式中,单水合物稳定至少一年,例如至少17个月。
在与第一方面相关的另一方面中,本发明提供依洛昔巴特的结晶单水合物,其中该结晶形式通过以下来制备:形成依洛昔巴特单醇化物,将单醇化物充分地转化为非溶剂合物并将非溶剂合物暴露于水蒸气。单醇化物可为甲醇化物、乙醇化物、1-丙醇化物、2-丙醇化物或这些醇的混合物。在某些实施方式中,在不形成单醇化物作为中间体的情形下不可形成单水合物。
本发明还包括其它结晶修饰物(包括结晶修饰物i及结晶修饰物iv)以及用于制备这些结晶修饰物的中间体。
本发明进一步提供治疗本文所描述病症的方法及本文所描述的结晶修饰物在治疗本文所描述病症及制备用于治疗本文所描述病症的药剂中的用途。
附图说明
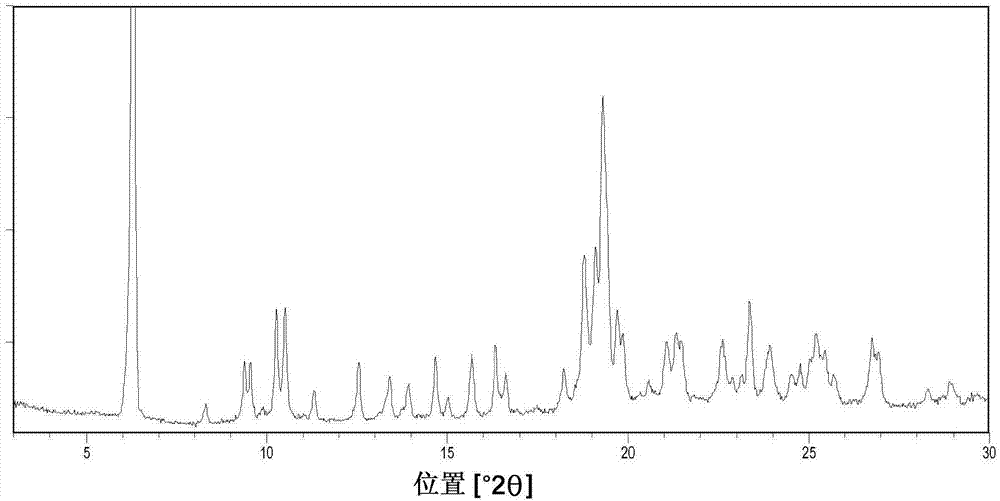
图1显示结晶修饰物iv的x射线粉末衍射图。
图2显示结晶修饰物etoh-1的x射线粉末衍射图。
图3显示结晶修饰物etoh-1(实线,底部)与iv(虚线,顶部)的x射线粉末衍射图间的比较。
图4显示结晶修饰物i的x射线粉末衍射图。
图5显示结晶修饰物iv在tga分析前(底部)及后(顶部)的x射线粉末衍射图。
图6显示由甲醇、乙醇、1-丙醇及2-丙醇获得的结晶修饰物iv的x射线粉末衍射图(来自底部)。
图7显示自甲醇、乙醇、1-丙醇及2-丙醇获得的结晶修饰物meoh-1、etoh-1、1-proh-1及2-proh-1的x射线粉末衍射图(来自底部)。
图8显示结晶修饰物meoh-1的x射线粉末衍射图。
图9显示结晶修饰物1-proh-1的x射线粉末衍射图。
图10显示结晶修饰物2-proh-1的x射线粉末衍射图。
图11显示结晶修饰物iv的dvs质量改变图形。该两个曲线显示%rh改变(右侧y轴)及试样重量%反应(左侧y轴)。在图表的最左侧显示预干燥步骤。
图12a及图12b显示水吸收随结晶修饰物iv的%rh变化的图形。图12a中所使用的试样由以实验室规模产生的物质获得,且图12b中所使用的试样由以中间工厂规模产生的gmp物质获得。
图13显示结晶修饰物i的dvs质量改变图形。该两个曲线显示%rh改变(右侧y轴)及试样重量%反应(左侧y轴)。在图表的最左侧显示预干燥步骤。
图14显示水吸收随结晶修饰物i的%rh变化的图形。
图15显示在略微不交叉的偏振器与使用10倍物镜的获得的结晶修饰物iv的显微图。
图16显示在略微不交叉的偏振器与使用10倍物镜的间拍下的结晶修饰物i的显微图。
图17显示结晶修饰物i及结晶修饰物iv的高分辨率x射线粉末衍射图。
图18显示结晶修饰物i、包含结晶修饰物i的片剂及安慰剂片剂的高分辨率x射线粉末衍射图。
图19显示结晶修饰物i、于40℃、75%相对湿度下储存8周后的包含结晶修饰物i的片剂及安慰剂片剂的高分辨率x射线粉末衍射图。
图20显示结晶修饰物iv、包含结晶修饰物iv的片剂及安慰剂片剂的高分辨率x射线粉末衍射图。
图21显示结晶修饰物iv、于40℃、75%相对湿度下储存8周后的包含结晶修饰物iv的片剂及安慰剂片剂的高分辨率x射线粉末衍射图。
具体实施方式
在第一方面中,本发明涉及依洛昔巴特的结晶修饰物iv。已惊奇地发现,可自初始认为最稳定的干燥形式(结晶修饰物i)起始获得此极稳定的依洛昔巴特的结晶修饰物。在i期及ii期临床试验中使用结晶修饰物i作为药物物质。当在约0℃至70℃之间(例如在约0℃至25℃之间)的温度下在乙醇或乙醇与水的混合物中浆液化此结晶修饰物i时,逐渐获得另一结晶修饰物,即乙醇溶剂合物etoh-1。已证实此溶剂合物为单乙醇化物。在(例如)减压及升高的温度下干燥此溶剂合物后,etoh-1失去其溶剂合物分子且变为部分结晶的非溶剂合物。当随后将非溶剂合物暴露于来自空气的水分时,其易于吸收一当量水。在该两相转化期间,或多或少地保存结晶结构。发现所得单水合物(在下文称作结晶修饰物iv)在(例如)环境开放条件下可稳定的储存至少上达至17个月。另外,此结晶修饰物较结晶修饰物i及依洛昔巴特的其它较少结晶形式具有更好的热力学稳定性及更高更一致的结晶度。
此后发现依洛昔巴特在室温下在其它醇(例如甲醇、1-丙醇及2-丙醇)或醇与水的50:50体积混合物中的表现类似。在这些条件下,可自浆液获得与etoh-1大体上等结构的溶剂合物meoh-1、1-proh-1及2-proh-1。由此形成的醇溶剂合物的表现与etoh-1的类似的处在于,其在其开始失去其溶剂合物分子时形成中间体,且然后当充分地蒸发掉醇时,其吸收水并转化为修饰物iv。图6显示自不同醇获得的修饰物iv的x射线粉末衍射数据。
此稳定结晶修饰物iv的分离不那么直截。尽管结晶修饰物iv涉及单水合物,但其不可直接自粗制依洛昔巴特或结晶修饰物i获得,因为当在水与醇的混合物中搅拌这些时,形成醇溶剂合物(meoh-1、etoh-1、1-proh-1或2-proh-1)。相信醇溶剂合物在这些条件下为热力学上更稳定的结晶修饰物。有趣地,如果不首先通过(例如)干燥自溶剂合物的结晶结构去除醇分子,则醇溶剂合物即使在暴露于100%相对湿度时还不能自发地转化为结晶修饰物iv。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.3±0.2及/或19.4±0.2处具有至少特定峰的x射线粉末衍射(xrpd)图案的结晶修饰物iv。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.3±0.2及19.4±0.2处具有特定峰且具有一个或多个以下特征峰的xrpd图案的结晶修饰物iv:10.2±0.2、10.5±0.2、9.4±0.2、9.5±0.2、12.5±0.2、14.6±0.2、15.6±0.2及23.3±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特定峰的xrpd图案的结晶修饰物iv:6.3±0.2、19.4±0.2、10.2±0.2、10.5±0.2、9.4±0.2及9.5±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物iv:6.3±0.2、19.4±0.2、10.2±0.2、10.5±0.2、9.4±0.2、9.5±0.2、12.5±0.2、14.6±0.2、15.6±0.2、23.3±0.2及一个或多个以下位置:8.3±0.2、11.3±0.2、13.4±0.2、13.9±0.2、16.3±0.2、16.6±0.2、18.2±0.2、18.8±0.2、19.1±0.2、19.3±0.2、19.7±0.2、19.8±0.2、20.5±0.2、21.0±0.2、21.3±0.2、21.4±0.2、22.6±0.2、22.9±0.2、23.1±0.2、23.9±0.2、24.5±0.2、24.7±0.2、25.0±0.2、25.2±0.2、25.4±0.2、25.7±0.2、26.7±0.2、26.9±0.2、28.3±0.2及28.9±0.2。
根据一个实施方式,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物iv:6.3±0.2、8.3±0.2、9.4±0.2、9.5±0.2、10.2±0.2、10.5±0.2、11.3±0.2、12.5±0.2、13.4±0.2、13.9±0.2、14.6±0.2、15.6±0.2、16.3±0.2、16.6±0.2、18.2±0.2、18.8±0.2、19.1±0.2、19.3±0.2、19.4±0.2、19.7±0.2、19.8±0.2、20.5±0.2、21.0±0.2、21.3±0.2、21.4±0.2、22.6±0.2、22.9±0.2、23.1±0.2、23.3±0.2、23.9±0.2、24.5±0.2、24.7±0.2、25.0±0.2、25.2±0.2、25.4±0.2、25.7±0.2、26.7±0.2、26.9±0.2、28.3±0.2及28.9±0.2。
在又一实施方式中,本发明涉及具有基本上如图1中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物iv。
在第二方面中,本发明涉及依洛昔巴特的结晶修饰物etoh-1。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.1±0.2及18.9±0.2处具有特征峰或在°2θ位置6.1±0.2及18.9±0.2处具有特征峰且具有一或多个以下特征峰的xrpd图案的结晶修饰物etoh-1:10.1±0.2、14.5±0.2、18.4±0.2、19.1±0.2、20.7±0.2、10.4±0.2、13.1±0.2及11.1±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物etoh-1:6.1±0.2、18.9±0.2、10.1±0.2、14.5±0.2、18.4±0.2、19.1±0.2、20.7±0.2、10.4±0.2、13.1±0.2、11.1±0.2及一或多个以下位置:8.0±0.2、9.3±0.2、12.2±0.2、13.7±0.2、15.1±0.2、15.3±0.2、15.9±0.2、17.2±0.2、17.8±0.2、20.3±0.2、21.2±0.2、22.0±0.2、22.2±0.2、22.5±0.2、23.6±0.2、24.0±0.2、24.5±0.2、24.7±0.2、25.2±0.2及26.3±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物etoh-1:6.1±0.2、8.0±0.2、9.3±0.2、10.1±0.2、10.4±0.2、11.1±0.2、12.2±0.2、13.1±0.2、13.7±0.2、14.5±0.2、15.1±0.2、15.3±0.2、15.9±0.2、17.2±0.2、17.8±0.2、18.4±0.2、18.9±0.2、19.1±0.2、20.3±0.2、20.7±0.2、21.2±0.2、22.0±0.2、22.2±0.2、22.5±0.2、23.6±0.2、24.0±0.2、24.5±0.2、24.7±0.2、25.2±0.2及26.3±0.2。
在又一实施方式中,本发明涉及具有基本上如图2中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物etoh-1。
在第三方面中,本发明涉及依洛昔巴特的结晶修饰物meoh-1。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.2±0.2及18.9±0.2处具有特征峰或在°2θ位置6.2±0.2及18.9±0.2处具有特征峰且具有一或多个以下特征峰的xrpd图案的结晶修饰物meoh-1:10.1±0.2、14.6±0.2、18.6±0.2、19.1±0.2、22.2±0.2、24.7±0.2、12.3±0.2、13.3±0.2及16.1±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物meoh-1:6.2±0.2、18.9±0.2、10.1±0.2、14.6±0.2、18.6±0.2、19.1±0.2、22.2±0.2、24.7±0.2、12.3±0.2、13.3±0.2、16.1±0.2及一或多个以下位置:8.1±0.2、9.3±0.2、10.5±0.2、10.9±0.2、13.0±0.2、14.4±0.2、15.8±0.2、17.6±0.2、20.3±0.2、20.7±0.2、21.0±0.2、22.7±0.2、24.0±0.2、24.3±0.2及26.1±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物meoh-1:6.2±0.2、8.1±0.2、9.3±0.2、10.1±0.2、10.5±0.2、10.9±0.2、12.3±0.2、13.0±0.2、13.3±0.2、14.4±0.2、14.6±0.2、15.8±0.2、16.1±0.2、17.6±0.2、18.6±0.2、18.9±0.2、19.1±0.2、20.3±0.2、20.7±0.2、21.0±0.2、22.2±0.2、22.7±0.2、24.0±0.2、24.3±0.2、24.7±0.2及26.1±0.2。
在又一实施方式中,本发明涉及具有基本上如图8中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物meoh-1。
在第四方面中,本发明涉及依洛昔巴特的结晶修饰物1-proh-1。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.1±0.2及19.0±0.2处具有特征峰或在°2θ位置6.1±0.2及19.0±0.2处具有特征峰且具有一或多个以下特征峰的xrpd图案的结晶修饰物1-proh-1:10.0±0.2、14.4±0.2、18.3±0.2、18.8±0.2、20.5±0.2、10.3±0.2、13.0±0.2及11.0±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物1-proh-1:6.1±0.2、19.0±0.2、10.0±0.2、14.4±0.2、18.3±0.2、18.8±0.2、20.5±0.2、10.3±0.2、13.0±0.2、11.0±0.2及一或多个以下位置:7.9±0.2、9.2±0.2、12.1±0.2、13.6±0.2、15.0±0.2、15.3±0.2、15.8±0.2、17.1±0.2、17.6±0.2、20.2±0.2、21.1±0.2、21.9±0.2、22.1±0.2、22.4±0.2、23.5±0.2、23.8±0.2、24.3±0.2、24.5±0.2、25.4±0.2及26.2±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物1-proh-1:6.1±0.2、7.9±0.2、9.2±0.2、10.0±0.2、10.3±0.2、11.0±0.2、12.1±0.2、13.0±0.2、13.6±0.2、14.4±0.2、15.0±0.2、15.3±0.2、15.8±0.2、17.1±0.2、17.6±0.2、18.3±0.2、18.5±0.2、18.8±0.2、19.0±0.2、19.4±0.2、20.2±0.2、20.5±0.2、21.1±0.2、21.9±0.2、22.1±0.2、22.4±0.2、23.1±0.2、23.5±0.2、23.8±0.2、24.3±0.2、24.5±0.2、25.4±0.2、26.0±0.2及26.2±0.2。
在又一实施方式中,本发明涉及具有基本上如图9中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物1-proh-1。
在第五方面中,本发明涉及依洛昔巴特的结晶修饰物2-proh-1。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置6.1±0.2及19.0±0.2处具有特征峰或在°2θ位置6.1±0.2及19.0±0.2处具有特征峰且具有一或多个以下特征峰的xrpd图案的结晶修饰物2-proh-1:10.0±0.2、14.4±0.2、18.3±0.2、18.8±0.2、20.5±0.2、10.3±0.2、12.9±0.2及11.0±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物2-proh-1:6.1±0.2、19.0±0.2、10.0±0.2、14.4±0.2、18.3±0.2、18.8±0.2、20.5±0.2、10.3±0.2、12.9±0.2、11.0±0.2及一或多个以下位置:9.1±0.2、12.1±0.2、13.6±0.2、14.9±0.2、15.2±0.2、15.7±0.2、17.1±0.2、17.6±0.2、18.5±0.2、19.4±0.2、20.2±0.2、21.1±0.2、21.7±0.2、22.1±0.2、22.3±0.2、23.1±0.2、23.4±0.2、23.7±0.2、24.1±0.2、24.4±0.2、24.6±0.2、25.1±0.2、25.4±0.2、25.9±0.2、26.2±0.2、27.4±0.2及29.2±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物2-proh-1:6.1±0.2、9.1±0.2、10.0±0.2、10.3±0.2、11.0±0.2、12.1±0.2、12.9±0.2、13.6±0.2、14.4±0.2、14.9±0.2、15.2±0.2、15.7±0.2、17.1±0.2、17.6±0.2、18.2±0.2、18.5±0.2、18.9±0.2、19.0±0.2、19.4±0.2、20.2±0.2、20.5±0.2、21.1±0.2、21.7±0.2、22.1±0.2、22.3±0.2、23.1±0.2、23.4±0.2、23.7±0.2、24.1±0.2、24.4±0.2、24.6±0.2、25.0±0.2、25.4±0.2、25.9±0.2、26.2±0.2、27.4±0.2及29.2±0.2。
在又一实施方式中,本发明涉及具有基本上如图10中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物2-proh-1。
在第六方面中,本发明涉及依洛昔巴特的结晶修饰物i。
在一个实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置5.2±0.2及/或10.0±0.2处具有特征峰的xrpd图案的结晶修饰物i。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在°2θ位置5.2±0.2及10.0±0.2处具有特征峰且具有一或多个以下特征峰的xrpd图案的结晶修饰物i:4.9±0.2、6.0±0.2、7.6±0.2、10.5±0.2、11.3±0.2、18.8±0.2、20.4±0.2及22.9±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物i:5.2±0.2、10.0±0.2、4.9±0.2、6.0±0.2、7.6±0.2、10.5±0.2及11.3±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物i:5.2±0.2、10.0±0.2、4.9±0.2、6.0±0.2、7.6±0.2、10.5±0.2、11.3±0.2、18.8±0.2、20.4±0.2、22.9±0.2及一或多个以下位置:3.1±0.2、4.4±0.2、7.4±0.2、7.8±0.2、8.2±0.2、12.4±0.2、13.3±0.2、13.5±0.2、14.6±0.2、14.9±0.2、16.0±0.2、16.6±0.2、16.9±0.2、17.2±0.2、17.7±0.2、18.0±0.2、18.3±0.2、19.2±0.2、19.4±0.2、20.1±0.2、20.7±0.2、20.9±0.2、21.1±0.2、21.4±0.2、21.8±0.2、22.0±0.2、22.3±0.2、23.4±0.2、24.0±0.2、24.5±0.2、24.8±0.2、26.4±0.2、27.1±0.2及27.8±0.2。
在另一实施方式中,本发明涉及具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案的结晶修饰物i:3.1±0.2、4.4±0.2、4.9±0.2、5.2±0.2、6.0±0.2、7.4±0.2、7.6±0.2、7.8±0.2、8.2±0.2、10.0±0.2、10.5±0.2、11.3±0.2、12.4±0.2、13.3±0.2、13.5±0.2、14.6±0.2、14.9±0.2、16.0±0.2、16.6±0.2、16.9±0.2、17.2±0.2、17.7±0.2、18.0±0.2、18.3±0.2、18.8±0.2、19.2±0.2、19.4±0.2、20.1±0.2、20.4±0.2、20.7±0.2、20.9±0.2、21.1±0.2、21.4±0.2、21.8±0.2、22.0±0.2、22.3±0.2、22.9±0.2、23.4±0.2、24.0±0.2、24.5±0.2、24.8±0.2、26.4±0.2、27.1±0.2及27.8±0.2。
在又一实施方式中,本发明涉及具有基本上如图4中所显示的利用cukα1辐射获得的xrpd图案的结晶修饰物i。
结晶修饰物iv的优点在于其在正常条件(21℃,10-30%相对湿度)下比结晶修饰物i及自甲醇、乙醇、1-丙醇或2-丙醇或自其中任一种醇类与水的混合物中获得的其它依洛昔巴特结晶修饰物在热力学上更稳定。此容许药物物质及药物配制剂的稳定且安全的制备制程。
依洛昔巴特的某些形式(例如依洛昔巴特的结晶修饰物i)含有非化学计量的水。在这些形式中,水的量可能变化(例如,随空气的相对湿度变化,或在不同批次之间变化)。相反,结晶修饰物iv是化学计量单水合物,即其每摩尔物质含有约一摩尔水(通常每摩尔物质0.9-1.1摩尔水,不包括吸附至晶体的表面的水)。此使得结晶修饰物iv在变化的相对湿度下具有更稳定的重量。
结晶修饰物iv是高度结晶单水合物,其可通过控制的转化过程经由乙醇溶剂合物etoh-1或经由等结构醇溶剂合物meoh-1、1-proh-1及2-proh-1产生。当蒸发乙醇且置换为水时,etoh-1的结晶结构仍然类似。此外,结晶修饰物iv的相对稳定的结晶度使得该化合物具有可再现的溶解性。此点对于欲用于药物制剂中的化合物而言特别重要,其中含有活性药物组分的每一片剂或胶囊应具有相同的药理学性质。因此,结晶修饰物iv比迄今所发现的其它依洛昔巴特结晶修饰物更有利于制备依洛昔巴特的药物配制剂。
结晶修饰物iv的又一优点在于,结晶修饰物i的结晶形态较平面(针形),与其相比,结晶修饰物iv的结晶形态较立体。此使得结晶修饰物iv在散装处置及配制方面具有有利性质。例如,可以减少或甚至不需要筛分材料,例如打破这些晶体,且可以在配制期间更易于与赋形剂混合。
在另一方面中,本发明涉及用于制备结晶修饰物iv的方法。此方法涉及由粗制或纯依洛昔巴特制备及分离结晶修饰物etoh-1或等结构醇溶剂合物meoh-1、1-proh-1及2-proh-1中的一种。在一个实施方式中,该方法包含以下步骤:
a)在器皿中制备依洛昔巴特处于醇或醇与水的混合物中的饱和溶液;
b)将过量依洛昔巴特添加至步骤a)的饱和溶液中以便获得浆液;
c)可选地在约5℃至25℃、优选20℃至25℃下将浆液的搅拌维持若干小时至多若干天或甚至一周或更长的时段;
d)回收在步骤c)中获得的固体,随后在真空中干燥固体直至充分地去除所有醇;及
e)将在步骤d)中获得的干燥固体暴露于来自空气的水分。
步骤a)中的粗制或纯起始材料是非结晶依洛昔巴特或依洛昔巴特的另一结晶修饰物。在某些实施方式中,依洛昔巴特基本上不含不同于水的溶剂。在优选实施方式中,起始材料是结晶修饰物i,其是依洛昔巴特的相对稳定的结晶修饰物。结晶修饰物i可如实验部分中所描述自粗制非结晶依洛昔巴特获得。其x射线粉末衍射图显示于图4中。
在某些实施方式中,步骤a)中所使用的依洛昔巴特饱和溶液除甲醇、乙醇、1-丙醇、2-丙醇及水以外不含任何溶剂,例如除甲醇、乙醇、1-丙醇、2-丙醇及水以外的溶剂少于0.5%w/w。若使用甲醇与水、乙醇与水、1-丙醇与水或2-丙醇与水的混合物,则甲醇、乙醇、1-丙醇或2-丙醇的量在某些实施方式中应为至少5%w/w。最优选地,溶剂为至少90%或甚至100%w/w甲醇、乙醇、1-丙醇或2-丙醇。
在步骤c)中获得的固体为结晶修饰物meoh-1、etoh-1、1-proh-1或2-proh-1。相信在甲醇、乙醇、1-丙醇或2-丙醇中或在甲醇、乙醇、1-丙醇或2-丙醇与水的混合物中,结晶修饰物meoh-1、etoh-1、1-proh-1或2-proh-1是热力学上最稳定的形式。因此,当在约5℃至25℃(例如20℃至25℃)(优选对于甲醇)下将步骤b)的悬浮液搅拌较长的时间段时,在该温度下,meoh-1、etoh-1、1-proh-1或2-proh-1将结晶。
结晶修饰物meoh-1是甲醇溶剂合物,结晶修饰物etoh-1是乙醇溶剂合物,结晶修饰物1-proh-1是1-丙醇溶剂合物,且结晶修饰物2-proh-1是2-丙醇溶剂合物。当在减压及升高的温度下干燥这些溶剂合物时,其失去其醇分子且变为非溶剂合物。为自醇溶剂合物形式完全转化为单水合物形式,必须干燥meoh-1、etoh-1、1-proh-1或2-proh-1,以便基本上去除包埋于晶体中的醇。优选地,在真空及升高的温度(例如约50℃或例如约65℃)下干燥固体。
当将非溶剂合物晶体暴露于来自空气的水分时,吸收水分子且形成单水合物结晶修饰物iv。在低至10%的相对湿度下发生水的吸收。对于可再生结果及高结晶度,优选在25℃下将脱水的晶体暴露于处于20-60%的相对湿度下的空气。借助于热重分析、差示扫描量热法、卡耳费雪滴定(karlfischertitration)及动态蒸气吸附分析,已显示结晶修饰物iv是单水合物。
可替换地,可通过将晶种晶体添加至依洛昔巴特处于甲醇、乙醇、1-丙醇或2-丙醇或甲醇、乙醇、1-丙醇或2-丙醇与水的混合物中的饱和溶液中制备结晶修饰物iv。因此,在另一实施方式中,该过程包含以下步骤:
a)在器皿中制备依洛昔巴特处于醇或醇与水的混合物中的过饱和溶液;
b)将晶种晶体添加至步骤a)的过饱和溶液中;
c)维持搅拌直至获得固体;
d)回收于步骤c)中获得的固体,随后在真空中干燥固体直至去除醇;及
e)将在步骤d)中获得的干燥固体暴露于来自空气的水分。
步骤a)中的粗制或纯起始材料涉及非结晶依洛昔巴特或依洛昔巴特的另一结晶修饰物,其在某些实施方式中不含不同于醇及水的溶剂。
在某些实施方式中,步骤a)中所使用的过饱和依洛昔巴特溶液不含除醇及水以外的任何溶剂,例如除醇及水的溶剂少于0.5%。若使用醇与水的混合物,则在某些实施方式中醇的量应为至少5%w/w。优选地,溶剂为甲醇、乙醇、1-丙醇或2-丙醇。
可通过以下制备过饱和溶液:将起始材料溶解于温甲醇、乙醇、1-丙醇或2-丙醇或甲醇、乙醇、1-丙醇或2-丙醇与水的温混合物中,且然后冷却所得溶液。温溶剂的初始温度优选地为约40℃至45℃,且然后将溶液冷却至如约25℃的温度。
晶种晶体应为结晶修饰物iv。晶种晶体的添加将加速meoh-1、etoh-1、1-proh-1或2-proh-1的形成及结晶。因此,步骤c)中的搅拌时间可显著更短,例如15小时或例如10小时。可在较低温度(例如5℃至10℃或例如0℃至5℃)下维持搅拌。
有趣地,尽管结晶修饰物iv是单水合物,但当在水与甲醇、乙醇、1-丙醇或2-丙醇的混合物中搅拌时,其不可由结晶修饰物i直接获得。在这种混合物中,结晶修饰物i分别转化为醇溶剂合物meoh-1、etoh-1、1-proh-1或2-proh-1,相信所有这些醇溶剂合物在这些条件下皆为比结晶修饰物i在热力学上更稳定的结晶修饰物。惊奇地,当随后将所形成的meoh-1、etoh-1、1-proh-1或2-proh-1暴露于100%相对湿度时,其仍不会转化为单水合物。此表明在水分子可能进入并改变结晶修饰物iv的结构前必须从结晶结构基本去除醇分子。
当在甲醇、乙醇、1-丙醇或2-丙醇中或在甲醇、乙醇、1-丙醇或2-丙醇与水的混合物中搅拌结晶修饰物iv时,其再次转化为meoh-1、etoh-1、1-proh-1或2-proh-1。据推测,仅在几分钟内发生此转化。此可能是结晶修饰物meoh-1、etoh-1、1-proh-1、2-proh-1及iv的xrpd图案之间的类似度较高(参见图3及图7)的结果。由于这些图案如此类似,故相信纵然不依靠该理论,在不存在溶解及随后的重结晶的情况下而是通过固态的重排还可发生转化。
依洛昔巴特是回肠胆汁酸转运体(ibat)抑制剂。回肠胆汁酸转运体(ibat)是自gi道再吸收胆汁酸的主要机制。部分或完全阻断该ibat机制将降低小肠壁、门静脉、肝实质、肝内胆及肝外胆包括胆囊中的胆汁酸浓度。可得益于部分或完全阻断ibat机制的疾病可为具有于血清及上述器官中的胆汁酸浓度过量的症状作为原发性病理生理学缺陷的疾病。
因此,在另一方面中,本发明还涉及依洛昔巴特的结晶修饰物iv,用于治疗中。
结晶修饰物iv可用于预防或治疗高胆固醇血症、异常血脂症、代谢综合征、肥胖、脂肪酸代谢失调、葡萄糖利用异常、涉及胰岛素抗性的失调、1型及2型糖尿病、肝病、包含ibat抑制剂化合物的疗法期间的腹泻、便秘(包括慢性便秘)(例如功能性便秘,包括慢性便秘及便秘型肠易激综合征(ibs-c))。便秘的治疗及预防描述于wo2004/089350中。
欲利用结晶修饰物iv治疗的其它可能疾病选自由以下组成的组中:肝实质、遗传型肝代谢失调、拜勒综合征(bylersyndrome)、胆汁酸(ba)合成的原发性缺陷(例如脑腱性黄色瘤病)、继发性缺陷(例如zellweger综合征(zellweger′ssyndrome))、新生儿肝炎、囊肿纤维变性(肝中的表现)、algs(alagille综合征)、渐进性家族性肝内胆汁郁积(pfic)、自体免疫肝炎、原发性胆汁性肝硬化(pbc)、肝纤维变性、非酒精性脂肪肝病、nafld/nash、门静脉高压症、一般胆汁郁积(例如药物引起或妊娠期间的黄疸性胆汁郁积)、肝内及肝外胆汁郁积(例如遗传型胆汁郁积,例如pfic1)、原发性硬化性胆管炎(psc)、胆结石及胆总管结石、引起胆树(biliarytree)阻塞的恶性肿瘤、由胆汁郁积/黄疸引起的症状(划伤、搔痒)、胰腺炎、导致渐进性胆汁郁积的慢性自体免疫肝病、胆汁郁积性肝病的搔痒及与高脂血症病症相关的疾病状态。
欲利用结晶修饰物iv治疗的其它疾病涉及选自由以下组成的组中:肝失调及与其相关的病症、脂肪肝、肝脂肪变性、非酒精性脂肪性肝炎(nash)、酒精性肝炎、急性脂肪肝、妊娠脂肪肝、药物诱导性肝炎、铁超载失调、肝纤维变性、肝硬化、肝肿瘤、病毒性肝炎及与肝、胆道及胰腺的肿瘤及赘瘤相关的问题。
因此,在一个实施方式中,本发明涉及依洛昔巴特的结晶修饰物iv,其用于治疗及/或预防如上文所列出的疾病或失调(disorder)。
在另一实施方式中,本发明涉及依洛昔巴特的结晶修饰物iv用于制备用于治疗和/或预防如上文所列出的疾病或失调的药剂的用途。
在又一实施方式中,本发明涉及治疗及/或预防温血动物的如上文所列出的疾病或失调的方法,其包含给予需要该治疗和/或预防的温血动物有效量的依洛昔巴特的结晶修饰物iv。
本发明的另一方面涉及药物组合物,其包含有效量的结晶修饰物iv以及药用稀释剂或载体。
本发明的又一方面涉及结晶修饰物iv用于制备药物组合物的用途,包括混合结晶修饰物iv与药用稀释剂或载体。
该药物组合物可进一步包含至少一种其它活性物质,例如选自以下的活性物质:ibat抑制剂;肠内分泌肽或其增强剂;二肽基肽酶-iv抑制剂;双胍;肠促胰液素模拟物;噻唑啉酮(thiazolidinone);ppar激动剂;hmgco-a还原酶抑制剂;胆汁酸黏合剂;tgr5受体调节剂;普洛斯通(prostone)类化合物的成员;鸟苷酸环化酶c激动剂;5-ht4血清素激动剂;或任一这些活性物质的药用盐。这些组合的实例还描述于wo2012/064268中。
结晶修饰物iv通常将以每平方米体表面积5mg至5000mg范围内的单位剂量(即约0.1mg/kg至100mg/kg或0.01mg/kg至50mg/kg)给予温血动物,且此通常提供治疗有效的剂量。单位剂型(例如片剂或胶囊)通常将含有约1mg至250mg活性组分,例如约1mg至100mg或约5mg至50mg,例如约1mg至20mg。可以单一剂量或分成一个、两个、三个或更多个单位剂量来给予日剂量。ibat抑制剂经口给予的日剂量优选地在0.1mg至1000mg内,更优选为1mg至100mg,例如5mg至15mg。
治疗性或预防性治疗所需要的剂量将取决于给予途径、疾病的严重性、患者的年龄及重量以及主治医师在确定适于具体患者的个别方案及剂量时通常考虑的其它因素。
定义
术语“结晶修饰物”是指有机化合物的结晶固相。结晶修饰物可为溶剂合物或非溶剂合物。
术语“溶剂合物”是指有机化合物的结晶固相,其具有并入其结晶结构中的溶剂分子。“水合物”是其中溶剂为水的溶剂合物,而“混合溶剂合物”是含有大于一种溶剂的分子的溶剂合物。
术语“浆液”是指其中添加过量固体的饱和溶液,从而形成固体及饱和溶液的混合物(即“浆液”)。
如本文所使用,术语“治疗(treatment)”、“治疗(treat)”和“治疗中(treating)”指逆转、减轻、延迟如本文所描述的疾病或失调或其一或多种症状的发作或抑制其进展。在一些实施方式中,可在已发生一或多种症状后给予治疗。在其它实施方式中,可在不存在症状下给予治疗。例如,可在症状发作前(例如,鉴于症状的历史及/或鉴于遗传或其它易感性因素)向易感个别给予治疗。还可在已解决症状后继续治疗以(例如)阻止或延迟其复发。
当本文提及结晶化合物时,如通过x射线粉末衍射数据评价的结晶度优选地大于约70%,例如大于约80%,具体而言大于约90%,更具体而言大于约95%。在本发明的实施方式中,如通过x射线粉末衍射数据评价的结晶度大于约98%,优选地大于约99%,其中结晶度%指为结晶的总试样物质的重量%。
优选地,根据本发明的结晶修饰物基本上不含该化合物的其它结晶修饰物。优选地,所描述的依洛昔巴特结晶修饰物包括少于(例如)20重量%、15重量%、10重量%、5重量%、3重量%或具体而言少于1重量%的其它依洛昔巴特结晶修饰物。因此,优选地,所描述的依洛昔巴特结晶修饰物的纯度>80%、>85%、>90%、>95%、>97%或具体而言>99%。
现将通过以下实施例描述本发明,这些实施例并非在任一方面限制本发明。所有引用文件及参考文献皆以引用方式并入。
缩写
cr.mod.结晶修饰物
etoh乙醇
h小时
hdpe高密度聚乙烯
ldpe低密度聚乙烯
meoh甲醇
min.分钟
1-proh1-丙醇
2-proh2-丙醇
实验方法
x射线粉末衍射(xrpd)分析
在玛瑙研钵中轻轻地研磨干燥试样(若需要),且然后涂抹于试样架上。将浆液试样以湿的形式添加至试样架中且实施湿式及干式分析。使用配备有x′探测器或pixcel检测器的panalyticalx′pertpro衍射仪在切割硅零背景支架(siliconzerobackgroundholder,zbh)上或在多孔氧化铝过滤器试样架上收集xrpd数据。在分析期间旋转试样且使用cu辐射。使用以下实验设置:
管电压及电流:40kv,50ma
波长α1(cukα1):
波长α2(cukα2):
波长α1及α2平均值(cukα):
起始角度[2θ]:1-4°
终止角度[2θ]:30-40°
分析时间:50s(“1min扫描”),125s(“2min扫描”),192s(“3min扫描”),397s(“6min扫描”),780s(“13min扫描”),1020s(“17min扫描”),4560s(“1h扫描”)除非另有指示,否则当自xrpd数据计算峰位置时,首先自来自cukα2的贡献除去数据且然后针对内部标准物(al2o3)进行校正。
本领域已知可获得具有一或多种取决于量测条件(例如所使用的设备、试样制备或机器)的量测误差的x射线粉末衍射图案。具体而言,通常已知xrpd图案中的强度可端视量测条件及试样制备而有所波动。例如,熟习xrpd技术者应认识到,峰的相对强度可根据所测试试样的定向及所用仪器的类型与设置而有所变化。熟习此项技术者还应认识到,反射位置可受试样位于衍射仪中的精确高度及衍射仪的零校准影响。试样的表面平面性还可具有小的效应。因此,本领域技术人员应了解,本文所呈现的衍射图案不应视为绝对的且提供与本文所公开了者实质上相同的粉末衍射图案的任一结晶形式属本发明的范围内(对于其它信息,参见r.jenkins及r.l.snyder,“introductiontox-raypowderdiffractometry”,johnwiley&sons,1996)。
热重分析(tga)
将约1-5mg试样添加至配衡铂杯中,然后将其置于perkin-elmerpyris1tga分析仪的称重位置中。升高炉子并记录试样的起始重量。然后,开始加热程序。以10℃/min的速率加热试样,在25℃开始且在90-300℃结束,端视可达到的恒定温度而定。在分析期间利用干燥氮气吹扫试样。
动态蒸气吸附(dvs)
将约15mg至20mg试样称重至石英容器中,然后通过暴露于放射性来源使该试样释放静电。然后,将石英容器置于surfacemeasurementssystemltddvsadvantage仪器中。利用干燥氮气干燥试样直至达到低于0.002%/分钟的dm/dt。以dm/dt模式使用5分钟的dm/dt窗口、10分钟的最小阶段时间及360分钟的最大阶段时间运行仪器。然后,使用上述d/m/dt模式参数使试样经受两个连续的吸附-去吸附循环,且每一循环涉及自0%–95%-0%相对湿度(%rh)运行。一个循环由20个步骤组成,彼等介于0至90%rh间的步骤各自涉及以10%rh实施。
差示扫描量热法(dsc)
将约2mg试样称重至利用铝盖非气密性密封的铝dsc盘(密封盘)中。然后,将试样装载至于30℃下冷却及保持的perkin-elmerdiamonddsc中。在获得充分稳定的热流反应后,以5℃/min的扫描速率将试样加热至150℃且监测所得热流反应。使用氮吹扫来防止试样在加热期间发生热诱导的氧化且还用来减少经由试样的热滞后从而增加仪器敏感度。分析前,使用铟参考标准在温度及热流方面校准仪器。
对于cryo-dsc实验,冷却perkin-elmerdiamonddsc并保持在5℃下,且然后,以10℃/分钟的扫描速率自5℃至200℃分析试样。
可如wo02/50051中所描述制备起始材料1,1-二氧基-3,3-二丁基-5-苯基-7-甲硫基-8-(n-{(r)-1'-苯基-1'-[n'-(叔丁氧基羰基甲基)氨基甲酰基]甲基}氨基甲酰基甲氧基)-2,3,4,5-四氢-1,5-苯并噻庚因。
实施例
实施例1
结晶修饰物i的制备
将甲苯(11.78l)在搅拌下填装至20l圆底烧瓶中且添加1,1-二氧基-3,3-二丁基-5-苯基-7-甲硫基-8-(n-{(r)-1'-苯基-1'-[n'-(叔丁氧基羰基甲基)氨基甲酰基]甲基}氨基甲酰基甲氧基)-2,3,4,5-四氢-1,5-苯并噻庚因(2.94kg)。在25-30℃下将甲酸(4.42l)添加至反应物质中。使温度升高至115-120℃并搅拌6小时。通过hplc监测反应以确保反应物质中剩余不超过1%的起始材料。使反应物质冷却至40-43℃。在搅拌的同时添加纯化水(11.78l)。使反应物质进一步冷却至25-30℃并搅拌15min。
分离各层并通过硅藻土床(0.5kg于3l甲苯中)过滤有机层并收集滤液。用甲苯(5.9l)洗涤硅藻土床,合并滤液并在真空下在38-40℃下浓缩。然后,使反应物质冷却至25-30℃以获得固体。
将乙醇(3.7l)在搅拌下装填至清洁圆底烧瓶中,并添加在前述步骤中获得的固体。将反应物质加热至40-43℃并在此温度下搅拌30min。然后,经30min的时段使反应物质冷却至25-30℃,且然后经2h的时段进一步冷却至3-5℃,随后在此温度下搅拌14h。将乙醇(3.7l)在搅拌下装填至反应物质中,同时使温度维持在0-5℃下,且然后将反应物质在此温度下搅拌1h。然后,过滤该物质并用乙醇(1.47l)洗涤,且真空干燥30min。将该物质在真空盘式干燥器中在37-40℃下在氮气氛下干燥24h。将该物质在氮气氛下放置于清洁双ldpe袋中并储存于清洁hdpe桶中。产量为1.56kg。
结晶修饰物i具有利用cukα1辐射获得且在以下°2θ位置处具有特征峰的xrpd图案:3.1±0.2、4.4±0.2、4.9±0.2、5.2±0.2、6.0±0.2、7.4±0.2、7.6±0.2、7.8±0.2、8.2±0.2、10.0±0.2、10.5±0.2、11.3±0.2、12.4±0.2、13.3±0.2、13.5±0.2、14.6±0.2、14.9±0.2、16.0±0.2、16.6±0.2、16.9±0.2、17.2±0.2、17.7±0.2、18.0±0.2、18.3±0.2、18.8±0.2、19.2±0.2、19.4±0.2、20.1±0.2、20.4±0.2、20.7±0.2、20.9±0.2、21.1±0.2、21.4±0.2、21.8±0.2、22.0±0.2、22.3±0.2、22.9±0.2、23.4±0.2、24.0±0.2、24.5±0.2、24.8±0.2、26.4±0.2、27.1±0.2及27.8±0.2。x射线粉末衍射图显示在图4中。
实施例2
结晶修饰物iv经由etoh-1的制备
在21℃下将依洛昔巴特结晶修饰物i(60mg)添加至乙醇(1.0ml)中及乙醇及水的混合物(0.25+0.75ml)中,以便产生浆液。然后,将搅拌棒添加至每一器皿中并闭合器皿。将器皿在21℃下充分搅拌一周。利用pasteur移液管取每一实验器皿中的固体残余物试样至切割硅零背景支架中,并利用两次连续的1分钟的xrpd扫描自1°至40°的2θ分析试样。此后,实施一或多个略微较长(3分钟及12秒)的xrpd分析直至获得两个连续且相同的xrpd衍射图。当已以此方式分析这些试样后,将其在开放实验室气氛中放置1天。在这些条件(约21℃及30%相对湿度)及小的试样大小下,自晶体蒸发乙醇分子且将其置换为水,由此产生结晶修饰物iv。
实施例3
结晶修饰物iv经由meoh-1的制备
将约80mg依洛昔巴特结晶修饰物iv添加至chromacol器皿中且然后添加1.0ml甲醇及搅拌蚤(stirringflea)。用卷压盖闭合器皿,在21℃下搅拌一天且然后取试样至切割硅零背景支架(zbh)中且在试样干燥时用xrpd进行重复分析。当目测干燥时,利用tga对其进行分析且然后使其吸收来自环境实验室气氛的水分,之后利用xrpd对其进行再分析。湿试样的xrpd数据显示于图8中且tga分析后的数据显示于图6中。
实施例4
结晶修饰物iv经由1-proh-1的制备
将约99.6mg依洛昔巴特结晶修饰物iv添加至chromacol器皿中且然后添加1.0ml1-丙醇及搅拌蚤。用卷压盖闭合器皿,在21℃下搅拌一天且然后取试样至切割硅零背景支架(zbh)中且在试样干燥时用xrpd进行重复分析。当目测干燥时,利用tga对其进行分析且然后使其吸收来自环境实验室气氛的水分,之后利用xrpd对其进行再分析。湿试样的xrpd数据显示于图9中且tga分析后的数据涉及于图6中给出。
实施例5
结晶修饰物iv经由2-proh-1的制备
将约103.5mg依洛昔巴特结晶修饰物iv添加至chromacol器皿中且然后添加1.0ml2-丙醇及搅拌蚤。用卷压盖闭合器皿,在21℃下搅拌一天且然后取试样至切割硅零背景支架(zbh)中且在试样干燥时用xrpd进行重复分析。当目测干燥时,利用tga对其进行分析且然后使其吸收来自环境实验室气氛的水分,之后利用xrpd对其进行再分析。湿试样的xrpd数据显示于图10中且tga分析后的数据涉及于图6中给出。
实施例6
结晶修饰物iv的制备
在氮气氛下在搅拌下将甲苯(145.9l)及1,1-二氧基-3,3-二丁基-5-苯基-7-甲硫基-8-(n-{(r)-1'-苯基-1'-[n'-(叔丁氧基羰基甲基)氨基甲酰基]甲基}氨基甲酰基甲氧基)-2,3,4,5-四氢-1,5-苯并噻庚因(7.295kg)装填至250-lglr中,且使反应物质冷却至3±2℃。经2-3h的时段,在3±2℃下将三氟乙酸(21.89l)缓慢添加至上述反应物质中。使反应物质的温度升高至25±5℃且在此温度下继续搅拌21h。通过hplc监测反应的进展。
使反应物质冷却至3±2℃并经30-40min的时段在3±2℃下在搅拌下添加纯化水(29.18l)。然后,使反应物质升温至25±5℃并在此温度下搅拌15min。使该物质沉降15min并分离各层。用水(3×29.18l)且然后用饱和盐水溶液(14.59l)洗涤有机层。在每次洗涤后,使该物质沉降15min的后分离各层。有机层通过不锈钢nutsche过滤器的硅藻土床(3.0kg硅藻土于17.0l甲苯中)过滤,并收集滤液。用甲苯(14.59l)洗涤硅藻土床。合并滤液并在低于40℃的温度下及真空(500-600mmhg)下浓缩至约7l至14l。
使上述物质冷却至25±5℃并经10-15min的时段添加正庚烷(72.95l)。将混合物在25±5℃下搅拌2h,然后过滤。用正庚烷(14.59l)洗涤过滤的固体并抽吸干燥约30min。
将上述粗制化合物在真空盘式干燥器中在38±2℃(500-600mmhg)下干燥10-12h。粗制重量:6.65kg。通过hplc得到的纯度:98.5%。
将无水乙醇(29.18l)装填至250l不锈钢反应器中并加热至43±2℃。将来自前述步骤的粗制产物(6.65kg)添加至预加热的乙醇中并在43±3℃下搅拌15min。然后,使所得溶液冷却至25±5℃并在此温度下搅拌1h。在此时期,溶液变浑浊。
利用结晶修饰物iv(2.0g)为晶种加至该物质中。然后,经2h的时段使该物质冷却至3±2℃,并在此温度下搅拌10h。过滤沉淀固体并用低温乙醇(1×3.65l)洗涤固体。将该物质抽吸干燥30min。然后,将该物质在真空盘干燥器中在25±5℃(500–600mmhg)下干燥24h且然后在63±2℃(约600mmhg)下干燥约50h。将经干燥产物储存在hdpe容器中。产量为5.31kg。
晶体吸收来自空气的水。测得水含量为2.70%。通过xrpd分析晶体且结果显示于图1中。
实施例7
结晶修饰物iv的热重分析
利用xrpd分析结晶修饰物iv的试样且利用tga检验水含量。结晶修饰物iv的重量损失起初较缓慢,但在约50℃下加速且在约80℃下结束。观察到重量损失为2.7%w/w。
可使用相同试样将该实验重复若干次,且结果相当类似。尽管在tga分析期间已自该试样蒸发水,但于tga分析的前及的后获得的x射线粉末衍射图涉及类似的(参见图5)。此指示水的吸收极迅速。另外,该实验显示该结晶修饰物极为稳定,因为在蒸发及再吸收水后维持结晶形状。
实施例8
结晶修饰物i的热重分析
利用xrpd分析结晶修饰物i的试样且利用tga检验水含量。在分析开始时立即出现重量损失且其在50℃至60℃下结束,从而指示此化合物中的水比结晶修饰物iv中的水结合的更松散。观察到重量损失为0.99%w/w。
实施例9
结晶修饰物iv的dsc分析
结晶修饰物iv在温度范围45℃至90℃内(56℃开始)展现吸热事件且在78℃下具有峰,且具有66.4j/g的焓。此事件是由于水的蒸发引起且对应于约2.9%w/w的水量。
在温度范围95℃至125℃内(103℃开始)观察到熔融峰,且在110℃下具有峰。
实施例10
结晶修饰物i的cryo-dsc分析
结晶修饰物i在温度范围15℃至85℃内(23℃开始)展现吸热事件且在56℃下具有峰,且具有23.2j/g的焓。此事件是由于水的蒸发引起且对应于约1.03%w/w的水量。
在温度范围110℃至145℃内(122℃开始)观察到熔融峰,且在131℃下具有峰。
实施例11
结晶修饰物iv的动态蒸气吸附分析
将结晶修饰物iv的试样称重至scientificinstruments动态蒸气吸附仪器的石英秤盘中。通过在试样上移动放射性同位素使该试样释放静电且然后将其放置至仪器中。通过吹扫干燥氮气干燥试样直至重量恒定且然后运行两个连续的吸附-去吸附循环。结晶修饰物iv在0%rh与10%rh之间吸收约2.45%水,且在10%rh与60%rh之间吸收额外0.36%至0.37%水。所得图表显示于图11中。
在图12a和图12b中,显示水吸收随%rh变化的图表。图12a中所使用的试样涉及自以实验室规模产生的物质获得,而图12b中所使用的试样涉及自以中间工厂规模产生的gmp物质获得。
实施例12
结晶修饰物i的动态蒸气吸附分析
将结晶修饰物i的试样称重至scientificinstruments动态蒸气吸附仪器的石英秤盘中。通过在试样上移动放射性同位素使该试样释放静电且然后将其放置至仪器中。通过吹扫干燥氮气干燥试样直至重量恒定且然后运行两个连续的吸附-去吸附循环。结晶修饰物i在0%rh与10%rh之间吸收约0.66%水,且在10%rh与60%rh之间吸收额外0.65%至0.69%水。所得图表显示于图13中。在图14中,显示水吸收随%rh变化的图表。
实施例13
结晶修饰物iv的稳定性测试
将一批结晶修饰物iv储存于封闭玻璃小瓶中并在20℃下及介于20%rh与60%rh之间保持17个月。xrpd数据指示该结晶形式在17个月后未改变。
实施例14
结晶修饰物iv的显微图
利用小刮勺将少量结晶修饰物iv放置于物镜载玻片上。添加一滴米格列醇(miglyol)并利用针状物使固体与液体充分混合,由此生成浆液。将盖玻片放置于浆液顶部并轻轻下推。然后,将物镜载玻片放置于nikonoptiphot-2偏振光显微镜的旋转台上。充分聚焦浆液的视野,且然后将光调节为科勒照明(
实施例15
结晶修饰物i的显微图
遵循实施例14中概述的程序但改为使用结晶修饰物i,获得显示于图16中的显微图。
实施例16
结晶修饰物etoh-1的热重分析
利用tga分析etoh-1试样的溶剂含量。观察到重量损失为约6%w/w,从而指示此结晶修饰物含有一摩尔乙醇。
实施例17
依洛昔巴特及包含结晶修饰物i或iv的片剂的高分辨率x射线粉末衍射图
量测方法:
在高辉度辐射设施‘spring-826b1’中实施x射线粉末衍射
检测器:r-axisv成像板检测器(制备商:rigaku)
辐射波长:
光束大小:100μm×100μm
试样与检测器之间的距离:420mm
用于量测的试样:包封于玻璃毛细管中
振动角度:80.0°
曝光时间:80秒
量测范围:3-15°(2θ)
量测温度:20℃
通过结晶修饰物i(在实施例1中获得)及结晶修饰物iv的spring-826b1实施x射线粉末衍射量测量。结果显示于图17中。
以显示于表1中的量混合各组分。利用制锭机(manestybetapress)在条件(重量:3.15-3.25g;高度:3.85mm)下使混合粉末形成为片剂,从而分别获得包含结晶修饰物i的片剂、包含结晶修饰物iv的片剂及安慰剂片剂。
表1
研磨包含结晶修饰物i的片剂以利用spring-826b1实施x射线粉末衍射测量。为鉴别不同于结晶修饰物i的衍射峰添加剂,利用spring-826b1以相同方式对安慰剂片剂实施x射线粉末衍射测量。发现包含结晶修饰物i的片剂的特征衍射峰(图18)。
将这些片剂于40℃、75%相对湿度的条件下储存8周。然后,利用spring-826b1对所储存片剂实施x射线粉末衍射测量(图19)。在x射线粉末衍射图的峰中未观察到改变,且发现片剂(结晶修饰物i)的特征衍射峰。
利用spring-826b1以与上文相同的方式对结晶修饰物iv实施x射线粉末衍射测量。发现包含结晶修饰物iv的片剂的特征衍射峰(图20)。
将这些片剂于40℃、75%相对湿度的条件下储存8周,但在x射线粉末衍射图的峰中未观察到改变;发现片剂(结晶修饰物iv)的特征衍射峰(图21)。
上述结果表明结晶修饰物iv可稳定存在于片剂中。
- 还没有人留言评论。精彩留言会获得点赞!