草铵膦的合成方法与流程
本发明涉及农药技术领域,特别涉及一种草铵膦的合成方法。
背景技术:
草铵膦(glufosinate-ammonium),化学名称为4-[羟基(甲基)膦酰基]-dl-丙氨酸,结构如下:
草铵膦(glufosinate)是一种高效低毒广谱触杀型有机磷类除草剂,它最早是由赫斯特公司(后归属于拜尔公司开发生产)。它是一种谷氨酰胺合成酶抑制剂,导致植物体内氮代谢紊乱、氨的过量积累、叶绿体解体,抑制其光合作用从而导致植物死亡。早些年,赫斯特公司成功将草铵膦的抗性基因导入了水稻、小麦、玉米等20多种作物中。草铵膦仅次于草甘膦成为世界第二大转基因作物耐受除草剂。草铵膦毒性低,较为安全,在土壤中易于降解,对作物安全,环境压力小。草铵膦的市场前景十分看好。
目前工业生产中草铵膦的主要合成方法是斯垂克法,同时也是比较成熟的传统方法,通过甲基亚磷酸酯与丙烯醛加成后,发生strecker反应、水解、纯化后得到高纯度的草铵膦。但其合成途径比较长,原料中用到了用到了氰化钠等剧毒原料,不利于环保,并且产生的三废处理比较复杂,成本高。
因此提供原料成本低廉、反应周期短、利于环保的制备草铵膦的方法具有重要意义。
目前有文献首次报道以γ-丁内酯为原料,经肟化,还原,氨基保护,氯化开环,与甲基亚磷酸二乙酯进行arbuzov反应可得到纯度较高的草铵膦,并且反应收率高,具有一定的工业化潜力。但其肟化反应使用到的叔丁醇钾和亚硝酸异丁酯,价格昂贵成本很高且不易得,还原反应涉及到氢气以及pd/c催化剂,其成本高且安全性有缺陷。其合成路线如下:
技术实现要素:
本发明提供一种制备草铵膦的方法,以低廉的γ-丁内酯为原料,反应收率较高,产品纯度高,条件温和且操作简单便捷,安全性好,符合绿色化工趋势,适于工业化生产。
本发明解决上述技术问题所采用的技术方案是:草铵膦的合成方法,包括有单溴取代,氨基化,氨基保护,氯化开环,阿尔布佐夫反应,酸化水解氨化,所述的单溴取代是γ-丁内酯ⅰ经催化剂与溴素发生α位单溴取代,减压蒸馏(b.p.120~125℃/1.6kpa)得到纯的中间体ⅱα-溴-γ-丁内酯,其中,所述催化剂为三溴化磷;所述的氨基化是α-溴-γ-丁内酯ⅱ与氨水发生氨基化反应,然后加盐酸回流得到中间体ⅲα-氨基-γ-丁内酯盐酸盐。
按上述方案:所述的氨基保护是将得到的中间体ⅲα-氨基-γ-丁内酯盐酸盐在溶剂存在下,加入适量的碱,与烷氧羰酰氯反应得到中间体ⅳ(2-氧代四氢呋喃-3-基)氨基甲酸乙酯。
按上述方案:所述的氯化开环是将得到的中间体ⅳ(2-氧代四氢呋喃-3-基)氨基甲酸乙酯与饱和的hcl乙醇溶液反应得到中间体ⅴ4-氯-2-乙氧羰基氨基丁酸乙酯;所述的阿尔布佐夫反应是将得到的中间体ⅴ4-氯-2-乙氧羰基氨基丁酸乙酯与甲基亚磷酸二乙酯经过arbuzov反应,得到中间体ⅵ4-乙氧基-4-甲基膦酰基-2-乙氧羰基氨基丁酸乙酯;所述的酸化水解氨化是将得到的中间体ⅵ4-乙氧基-4-甲基膦酰基-2-乙氧羰基氨基丁酸乙酯先经过盐酸水解,然后加氨水反应得到产物草铵膦铵盐。
按上述方案:所述的溴素与γ-丁内酯的摩尔比为1-2:1。
按上述方案:所述的溴素与γ-丁内酯的摩尔比为2:1。
按上述方案:所述的中间体ⅱ与氨水的摩尔比为1:3-9,中间体ⅱ与盐酸的摩尔比为1:1-2.5。
按上述方案:所述的中间体ⅱ与氨水的摩尔比为1:6,中间体ⅱ与盐酸的摩尔比为1:1.5。
按上述方案:所述的碱为na2co3或三乙胺。
按上述方案:所述的烷氧羰酰氯为氯甲酸乙酯、氯甲酸甲酯和苄氧甲酰氯中的任意一种。
按上述方案:所述的溶剂为二氯甲烷,以及水与四氢呋喃的混合液中的一种。
本发明所涉及的反应方程式为:
其中r1为c1-c2的烷基或bn。
本发明的有益效果在于:
1)以低廉的γ-丁内酯为原料,与溴素作用发生单溴取代,然后与氨水进行氨基化反应,所使用的原料便宜易得,反应条件温和,操作简捷,安全性高,放大生产可行,并且反应收率高,产品纯度高,大大降低了成本,适合工业化生产;
2)本发明使用便宜易得的溴素作为溴源,选择性单溴取代反应收率在88%以上,减压蒸馏后产品纯度高,溴代反应中γ-丁内酯既是原料又是溶剂,降低了成本与减少了后处理的步骤;
3)本发明氨基化反应生成中间体ⅲα-氨基-γ-丁内酯盐酸盐的条件温和,反应收率在70%以上,后处理简单且得到产物纯度很高;
4)本发明在氨基保护生成中间体ⅳ的反应中,使用了四氢呋喃与水的混合溶剂体系,且选择使用无机碱体系,反应收率在85%以上;
5)本发明在氯化开环生成中间体ⅴ的反应中,选用饱和hcl的乙醇溶液,该体系绿色环保,收率在65%以上。
附图说明
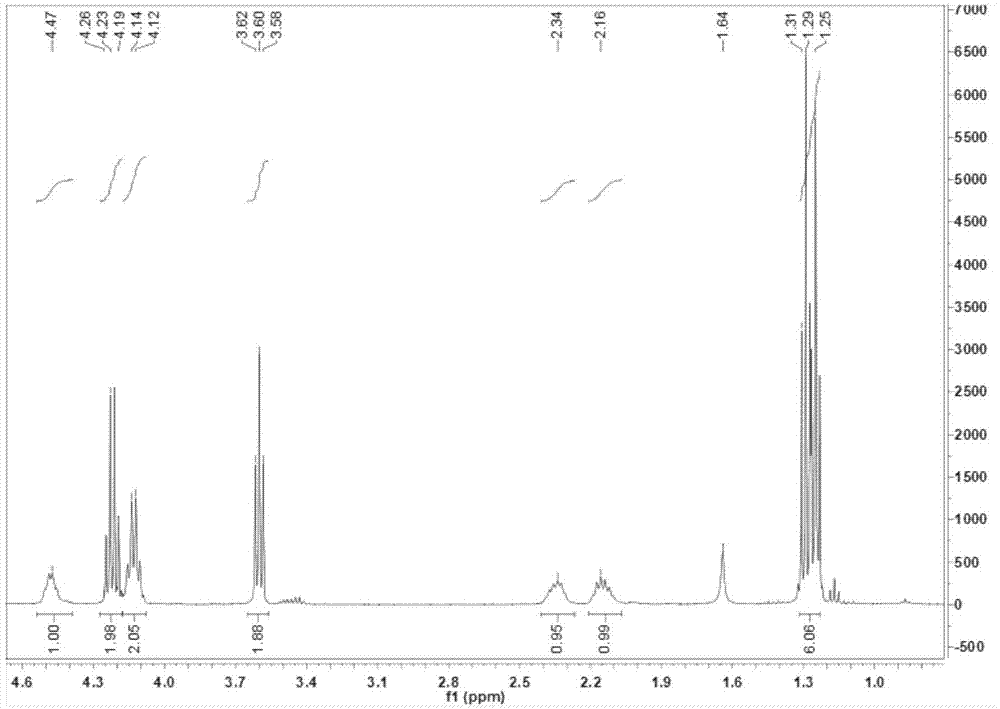
图1为本发明制备的式ⅴ化合物4-氯-2-乙氧羰基氨基丁酸乙酯的1h-nmr图;
图2为本发明制备的式ⅶ化合物2-氨基-4-(羟基甲基膦酰基)丁酸的1h-nmr图;
图3为本发明制备的式ⅶ化合物2-氨基-4-(羟基甲基膦酰基)丁酸的13c-nmr图;
图4为本发明制备的式ⅶ化合物2-氨基-4-(羟基甲基膦酰基)丁酸的31p-nmr图。
具体实施方式
下面通过具体实施例对本发明作进一步详细介绍:
实施例1:中间体ⅱα-溴-γ-丁内酯的制备方法:
冰浴下,在三口瓶中往500gγ-丁内酯和31g红磷的混合物中缓慢滴加330ml液溴,然后升温至70℃,继续滴加330ml液溴。滴加完毕后,升温至80℃反应3h。停止加热至冷却,持续吹入空气,将过量的液溴和生成的溴化氢吹走。然后再升温至80℃,缓慢滴加100ml水,等反应缓和后再加入700ml水,回流4h。冷却后分层,水层用二氯甲烷萃取,合并有机层,无水硫酸镁干燥。减压蒸去溶剂后得到粗产物(b.p.120~125℃/1.6kpa),减压蒸馏精制后,得淡黄色油状液体850g,收率88.7%。
实施例2:中间体ⅱα-溴-γ-丁内酯的制备方法:
冰浴下,在三口瓶中往500gγ-丁内酯和31g红磷的混合物中缓慢滴加330ml液溴,然后升温至70℃,继续滴加250ml液溴。滴加完毕后,升温至80℃反应3h。停止加热至冷却,持续吹入空气,将过量的液溴和生成的溴化氢吹走。然后再升温至80℃,缓慢滴加100ml水,等反应缓和后再加入750ml水,回流4h。冷却后分层,水层用二氯甲烷萃取,合并有机层,无水硫酸镁干燥。减压蒸去溶剂后得到粗产物,减压蒸馏精制,得淡黄色油状液体760g,收率80%。
实施例3:中间体ⅱα-溴-γ-丁内酯的制备方法:
冰浴下,在三口瓶中往500gγ-丁内酯和31g红磷的混合物中缓慢滴加330ml液溴,然后升温至70℃,继续滴加150ml液溴。滴加完毕后,升温至80℃反应3h。停止加热至冷却,持续吹入空气,将过量的液溴和生成的溴化氢吹走。然后再升温至80℃,缓慢滴加100ml水,等反应缓和后再加入750ml水,回流4h。冷却后分层,水层用二氯甲烷萃取,合并有机层,无水硫酸镁干燥。减压蒸去溶剂后得到粗产物,减压蒸馏精制,得淡黄色油状液体610g,收率64.2%。
1hnmr(600mhz,cdcl3)δ:4.48(td,1h),4.41(ddd,2h),2.84–2.74(m,1h),2.47(ddt,1h).
实施例1中,加入的溴素与γ-丁内酯的当量比为2,最终得到淡黄色油状液体850g,收率达88.7%;实施例2中,加入的溴素与γ-丁内酯的当量比为1.8,最终得到淡黄色油状液体760g,收率达80%;实施例3中,加入的溴素与γ-丁内酯的当量比为1.5,最终得到淡黄色油状液体610g,收率达64.2%。故当加入的溴素与γ-丁内酯的当量比为2,收率达88.7%,效果最好。
实施例4:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的实施例1的(下同)溴代产物缓慢地滴加到136ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸,在110℃下回流1h,抽滤得到39.4g的白色固体,收率50.3%。
实施例5:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的溴代产物缓慢地滴加到270ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸,在110℃下回流1h,抽滤得到48.25g的白色固体,收率70.6%。
实施例6:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的溴代产物缓慢地滴加到46ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸,在110℃下回流1h,抽滤得到18.5g的白色固体,收率10.2%。
实施例4中,氨水与α-溴-γ-丁内酯的当量比为3eq时,得到产物中间体ⅲ白色固体有39.4g,收率达50.3%;实施例5中,氨水与α-溴-γ-丁内酯的当量比为6eq时,得到产物中间体ⅲ白色固体有48.25g,收率达70.6%;实施例6中,氨水与α-溴-γ-丁内酯的当量比为1eq时,得到产物中间体ⅲ白色固体有18.5g,收率达10.2%.故当氨水与α-溴-γ-丁内酯的当量比为6eq时,收率达70.6%,效果最好。
实施例7:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的溴代产物缓慢地滴加到270ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸26ml,在110℃下回流1h,抽滤得到40.96g的白色固体,收率42.8%。
实施例8:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的溴代产物缓慢地滴加到270ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸39ml,在110℃下回流1h,抽滤得到46g的白色固体,收率68.3%。
实施例9:中间体ⅲα-氨基-γ-丁内酯盐酸盐的制备方法:
在冰浴条件下,将50g的溴代产物缓慢地滴加到270ml25wt%的氨水中,反应48h后,减压蒸馏除去溶剂,随后加浓盐酸65ml,在110℃下回流1h,抽滤得到36.17g的白色固体,收率40.3%。1hnmr(600mhz,dmso)δ8.89(s,2h),4.46–4.25(m,3h),2.59–2.52(m,1h),2.36–2.26(m,1h).
实施例7中,盐酸与α-溴-γ-丁内酯的当量比为1时,得到产物中间体ⅲ白色固体有40.96g,收率为42.8%;实施例8中,盐酸与α-溴-γ-丁内酯的当量比为1.5时,得到产物中间体ⅲ白色固体有46g,收率为68.3%;实施例9中,盐酸与α-溴-γ-丁内酯的当量比为2.0时,得到产物中间体ⅲ白色固体有36.17g,收率为40.3%;故当盐酸与α-溴-γ-丁内酯的当量比为1.5eq时,得到产物中间体ⅲ白色固体有46g,收率为68.3%,效果最好。
实施例10:中间体ⅳ的制备方法:
在冰浴条件下,取氨基化产物白色固体20g与100ml四氢呋喃和200ml水混合,往其中加入碳酸钠,再将19g的氯甲酸乙酯缓慢滴加进去反应半小时后,恢复至室温后继续反应2h,减压蒸馏除去溶剂,调节ph至酸性,用二氯甲烷萃取得到粗式ⅳ化合物,降温后有固体析出,抽滤得到无色油状液体13.08g。收率为87%。1hnmr(600mhz,cdcl3)δ:5.20(s,1h),4.47–4.12(m,5h),2.79(s,1h),2.27–2.15(m,1h),1.60(s,2h),0.87(s,1h).
实施例11:中间体ⅳ的制备方法:
在冰浴条件下,取氨基化产物白色固体20g与四氢呋喃和水混合,往其中加入碳酸钠,再将30g的氯甲酸苄酯缓慢滴加进去反应半小时后,恢复至室温后继续反应2h,减压蒸馏除去溶剂,调节ph至酸性,用二氯甲烷萃取得到粗的式ⅳ化合物,降温后有固体析出,抽滤得到产品无色油状液体13.54g,收率为90%。1hnmr(600mhz,cdcl3)δ:7.36~7.25(m,5h),5.34(s,1h),5.13(s,2h),4.48~4.21(m,3h),2.80~2.75(m,1h),2.27~2.18(m,1h).
实施例12:中间体ⅳ的制备方法:
在冰浴条件下,取氨基化产物白色固体20g与100ml四氢呋喃和200ml水混合,往其中加入碳酸钠,再将16.5g的氯甲酸甲酯缓慢滴加进去反应半小时后,恢复至室温后继续反应2h,减压蒸馏除去溶剂,调节ph至酸性,用二氯甲烷萃取得到粗的式ⅳ化合物,降温后有固体析出,抽滤得到无色油状液体9.0g。两步收率为60%。1hnmr(600mhz,cdcl3)δ:5.52(s,1h),4.49~4.23(m,3h),3.71(s,3h),2.79~2.70(m,1h),2.28–2.21(m,1h).
实施例13:中间体ⅳ的制备方法:
在冰浴条件下,取氨基化产物白色固体20g加入到含有20ml的三乙胺的ch2cl2(300ml),再将30g的氯甲酸苄酯缓慢滴加进去反应半小时后,恢复至室温后继续反应2h,减压蒸馏除去溶剂,调节ph至酸性,用二氯甲烷萃取得到粗的式ⅳ化合物,降温后有固体析出,抽滤得到产品无色油状液体3.0g。两步收率为20%。
实施例14:中间体ⅴ的制备方法:
取10g式ⅳ化合物,加入到200ml的耐压管中,随后将150ml的饱和的hcl乙醇溶液加入其中,升温至65℃反应12h后,旋蒸掉其中的乙醇溶剂,加入少量的水,在用乙酸乙酯萃取三遍,旋掉溶剂后,然后用石油醚洗涤,得到无色油状液体8.12g即式ⅴ化合物,收率在60%以上。1hnmr(400mhz,cdcl3)δ:5.29(s,1h),4.47(s,1h),4.27–4.18(m,2h),4.13(d,2h),3.60(t,2h),2.34(s,1h),2.16(s,1h),1.32–1.23(m,6h),1h-nmr如图1所示。
实施例15:中间体ⅵ的制备方法:
取10.0g如式ⅴ的中间体4与6.9g甲基亚磷酸二乙酯在150ml无水甲苯中混合,氮气保护下,回流反应10h。减压蒸馏去除甲苯和过剩甲基亚磷酸二乙酯,残余物溶于乙酸乙酯后,盐水洗涤3次,有机层干燥,减压蒸馏去除溶剂,得9.6g如式ⅵ的中间体5收率75.87%。1hnmr(400mhz,dmso-d6)δ:7.49(s,1h),4.17–3.97(m,6h),2.14~1.91(m,2h),1.65–1.07(m,13h).
实施例16:式ⅶ产物的制备方法:
称取10.0g如式ⅵ的中间体加入到100ml6mol/lhcl溶液中,回流8h。再用28%氨水将ph值调到9,继续回流6h,减压蒸馏后,用甲醇重结晶,得到白色草铵膦晶体5.2g,收率80.96%。1hnmr(400mhz,d2o)δ:3.67(dd,1h),2.04~1.87(m,2h),1.58–1.41(m,2h),1.14(d,3h).13cnmr(101mhz,d2o)δ:172.3,53.7(d,jc-p=14.1hz),25.9(d,jc-p=92.0hz),23.2(d,jc-p=2.3hz),14.1(d,jc-p=93.2hz).31pnmr(162mhz,d2o)δ:49.04,1h-nmr如图2所示,13c-nmr如图3所示,31p-nmr如图4所示。
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的实施例。
- 还没有人留言评论。精彩留言会获得点赞!