一种西他沙星中间体的制备方法与流程
本发明属于有机合成技术领域,具体是涉及一种西他沙星的中间体的制备方法。
背景技术:
西他沙星(sitafloxacin),化学名为(-)-7-[(7s)-7-氨基-5-氮杂螺[2.4]庚-5-基]-8-氯-6-氟-1-[(1r,2s)-2-氟-1-环丙基]-1,4-二氢-4-氧代-3-喹啉羧酸,是日本第一制药三共株式会社开发的广谱喹诺酮类抗菌药,2008年首次在日本上市。临床用其3/2水合物,用于治疗严重难治性细菌感染、复发性感染以及某些耐药菌感染。西他沙星能抑制细菌dna促旋酶和拓扑同工酶,对该两种酶的抑制作用较其它喹诺酮类药物要强。
西他沙星由于结构中含有一个顺式氟环丙胺基团,而具有良好的药代动力学特性,并可以减轻不良反应,其体外抗菌性较大多数同类药物明显增强。对革兰氏阴性菌、革兰氏阳性球菌以及压氧菌的抗菌活性是左氧氟沙星的4-32倍,对许多临床常见的耐喹诺酮类菌株也具备良好的杀菌作用。西他沙星口服吸收好,生物利用率大于70%,组织分布广,在中枢神经系统外的许多组织中的药物浓度均高于血清浓度。因此,西他沙星有望成为治疗呼吸道,泌尿生殖道、腹腔以及皮肤软组织等单一或者混合细菌感染的重要药物。
西他沙星结构式如式i所示:
西他沙星中间体ii,化学名为2-(3-氯-2,4,5-三氟-苯甲酰基)-3-乙氧基-丙酸乙酯,是一种化学合成西他沙星过程中的重要的中间体,其结构式如下所示:
现有参考文献(h.liuetal./europeanjournalofmedicinalchemistry86(2014)-628-638)合成路线如下:
现有技术的合成路线具有步骤相对较长,后处理繁琐,副反应多,产品收率低,成本高,污染大等缺点,寻找一条经济安全,绿色环保的合成路线具有重要意义。
技术实现要素:
本发明提供了一种西他沙星中间体的制备方法,属于化学药物制备工艺领域。
本发明是通过以3-氯-2,4,5-三氟苯甲酸作为起始原料,经过与氯化亚砜反应成酰氯,再与3-溴-2-乙氧基丙烯酸乙酯反应生成化合物即为本发明所述的西他沙星中间体ⅱ。
一种西他沙星中间体的制备方法,包括:低价金属或过渡金属存在下,3-氯-2,4,5-三氟苯甲酰氯与3-溴-2-乙氧基丙烯酸乙酯反应,得到所述的西他沙星中间体;所述西他沙星中间体结构如下:
作为优选,所述3-氯-2,4,5-三氟苯甲酰氯由3-氯-2,4,5-三氟苯甲酸与酰氯反应得到。反应过程如下:
具体包括如下步骤:
(1)起始原料3-氯-2,4,5-三氟苯甲酸(iii)以低级卤代烷烃作溶剂,低温下缓慢加入一种酰氯反应制备成中间体酰氯(iv);
(2)3-溴-2-乙氧基丙烯酸乙酯(v)以低级卤代烷烃作溶剂,加入低价金属或过渡金属,低温下缓慢滴加中间体酰氯(iv)溶液,滤掉不溶物,水洗,分成,干燥得到西他沙星中间体ⅱ(可以以溶液形式直接进行下步取代反应,也可以减压浓缩成液体)。
本发明中,所述低价卤代烷烃溶剂包括二氯甲烷、氯仿、二氯乙烷、1,1-二氯乙烷中的一种或多种。
本发明中,所述低价金属或者过渡金属包括镁、铜、锌、铁、钴的一种或多种。
本发明中,步骤(1)所述酰氯一般为氯化亚砜、草酰氯、硫酰氯、三光气、氯甲酸乙酯中的一种。
本发明中,步骤(1)和(2)所述低温一般为-20~10℃。
本发明中,作为优选,所述3-氯-2,4,5-三氟苯甲酸与酰氯的摩尔比为1:(1~2)。
本发明中,作为优选,所述3-氯-2,4,5-三氟苯甲酸与3-溴-2-乙氧基丙烯酸乙酯摩尔比为1:(1~2)。
本发明中,作为优选,3-氯-2,4,5-三氟苯甲酸与低价金属或过渡金属的摩尔比为1:(1~2)。
本发明中,作为优选,3-氯-2,4,5-三氟苯甲酸与酰氯反应完成后,55-60℃常压回收二氯甲烷,再减压蒸出多余的氯化亚砜,得到3-氯-2,4,5-三氟苯甲酰氯,然后再将回收的二氯甲烷将所述3-氯-2,4,5-三氟苯甲酰氯溶解,直接进入下一步反应。
作为优选,3-氯-2,4,5-三氟苯甲酸与酰氯反应时加入dmf。
作为优选,3-氯-2,4,5-三氟苯甲酸与酰氯反应时,采用在-20~10℃下,加入酰氯;3-氯-2,4,5-三氟苯甲酰氯与3-溴-2-乙氧基丙烯酸乙酯反应时,采用在-20~10℃下,加入3-氯-2,4,5-三氟苯甲酰氯。
本发明中,在没有特殊说明时,均是在室温下进行。
与现有技术相比,本发明的西他沙星中间体ⅱ的制备方法,合成路线较为简捷,反应条件较为温和,后处理简单,能耗低,溶剂回收率高,同时采用原料和试剂价格便宜易得到。
本发明提供的制备方法,收率均在96%以上,经过简单常规后处理,得到的产品纯度均在99%以上,适于工业化大量生产,具有较好的市场前景。
说明书附图
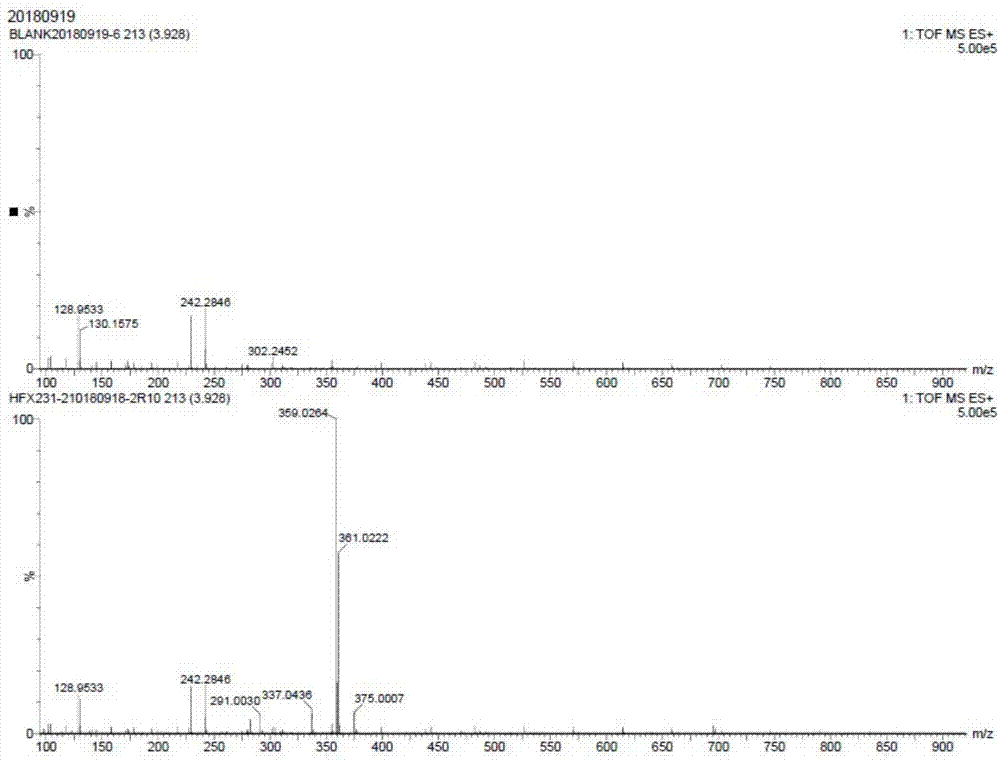
图1为实施例1制备得到的西他沙星中间体ⅱ的质谱数据。
具体实施方式
下面通过具体实施例对发明作进一步说明。
实施例1:
500ml四口瓶中加入(21g,0.10mol)3-氯-2,4,5-三氟苯甲酸、210ml二氯甲烷和1mldmf,搅拌至溶清,降温至0-10℃,缓慢滴加入(13.1g,0.11mol)氯化亚砜,滴加完毕,转移至室温下反应3h,55-60℃常压蒸出二氯甲烷,再减压蒸出多余的氯化亚砜,得到中间体酰氯(iv),反应瓶中加入100ml蒸出的二氯甲烷,搅拌,配成溶液待用。
500ml反应瓶中加入(23.4g,0.105mol)3-溴-2-乙氧基丙烯酸乙酯(v)、100ml二氯甲烷,搅拌均匀,降温至0-10℃,加入(6.8g,0.105mol)锌粉,搅拌0.5h,缓慢滴加中间体酰氯(iv)溶液,滴加完毕,转移室温下反应2h,过滤,滤液中加入100ml水,搅拌,静置,分层,分出有机相,加入无水硫酸钠干燥,过滤,滤液先55-60℃常压回收二氯甲烷,然后再20-30℃减压浓缩得到32.6g微黄色液体西他沙星中间体ⅱ,收率为97.2%,纯度为90.6%。质谱数据为:359.0264(+na),如图1所示。
利用该化合物制备对比文件(europeanjournalofmedicinalchemistry86(2014)628e638,参见p637:4.2.2.以及4.2.2.3.)中的化合物7c(ethyl8-chloro-6,7-difluoro-1-[(1r,2s)-2-fluorocyclopropyl]-4-oxo-1,4-dihydroquinoline-3-carboxylate7),检测得到化合物7c的核磁数据与该对比文件中公开的数据完全一致,进一步证明了本发明方法的可行性。
实施例2:
500ml四口瓶中加入(21g,0.10mol)3-氯-2,4,5-三氟苯甲酸、210ml二氯甲烷和1mldmf,搅拌至溶清,降温至0-10℃,缓慢滴加入(13.1g,0.11mol)氯化亚砜,滴加完毕,转移至室温下反应4h,得到中间体酰氯(iv)溶液,待用。
500ml反应瓶中加入(23.4g,0.105mol)3-溴-2-乙氧基丙烯酸乙酯(v)、100ml二氯甲烷,搅拌均匀,降温至0-10℃,加入(7.2g,0.11mol)锌粉,搅拌1h,缓慢滴加中间体酰氯(iv)溶液,滴加完毕,转移室温下反应3h,过滤,滤液中加入150ml水,搅拌,静置,分层,分出有机相,加入无水硫酸钠干燥,过滤,滤液先55-60℃常压回收二氯甲烷,然后再20-30℃减压浓缩得到32.8g微黄色液体西他沙星中间体ⅱ,收率为97.6%,纯度为90.3%。
实施例3:
500ml四口瓶中加入(21g,0.10mol)3-氯-2,4,5-三氟苯甲酸、210ml二氯甲烷和1mldmf,搅拌至溶清,降温至0-10℃,缓慢滴加入(13.4g,0.105mol)草酰氯,滴加完毕,转移至室温下反应3h,得到中间体酰氯(iv)溶液,待用。
500ml反应瓶中加入(23.4g,0.105mol)3-溴-2-乙氧基丙烯酸乙酯(v)、100ml二氯甲烷,搅拌均匀,降温至0-10℃,加入(7.2g,0.11mol)铁粉,搅拌1h,缓慢滴加中间体酰氯(iv)溶液,滴加完毕,转移室温下反应2h,过滤,滤液中加入150ml水,搅拌,静置,分层,分出有机相,加入无水硫酸钠干燥,过滤,滤液先55-60℃常压回收二氯甲烷,然后再20-30℃减压浓缩得到32.5g微黄色液体西他沙星中间体ⅱ,收率为96.7%,纯度为89.9%。
- 还没有人留言评论。精彩留言会获得点赞!