一种具有高let-7b成熟体表达量的单倍型鸡成肌细胞的构建方法与流程
本发明涉及生物基因工程领域,特别涉及一种单倍型鸡成肌细胞的构建方法。
背景技术:
microrna(mirna)是一类约有22个左右核苷酸的内源性非编码单链rna。随着分子生物研究技术与方法的快速发展,研究人员发现了越来越多的mirna。mirna是由一段长度为70~80个核苷酸的且包含一段发夹结构的mirna前体(pre-mirna)剪切后生成。mirna通过与其靶标基因mrna的3’端非编码区域(3-untranslatedregion,3’utr)互补导致该mrna的翻译受到抑制实现对基因的调控。mirna虽然不直接编码蛋白质,但其编码的rna在生物的整个生命过程中发挥着重要作用。目前发现mirnas能够参与许多调节途径,包括骨骼肌发育、病毒防御机制、造血过程、器官形成分化、细胞增殖和死亡、脂肪代谢等。
近些年来,随着基因组学和全基因组关联分析等技术的迅速发展,mirna已然成为国内外动物遗传育种研究中的热门领域。在国内,金晶等利用高通量测序技术对荷斯坦牛泌乳期高乳品质奶牛(h)和泌乳期低乳品质奶牛(l)乳腺组织进行了mirna测序,通过差异表达分析筛选组间差异mirnas,获得56个差异表达mirna(p<0.05),并对差异表达mirna进行靶基因预测,经过对靶基因筛选,发现了4个已报道与乳蛋白、乳脂紧密相关的功能基因:csn3、scd、lalba和dgat2。靶基因聚集的生物学功能多数参与了蛋白质和脂肪代谢,乳腺发育和分化,以及免疫功能。凌英会等对山羊肌肉组织进行solexa测序并进行生物信息学分析,共鉴定出517个物种间保守的和2个山羊基因组特有的mirna。本课题组前期通过靶标预测软件预测并通过双荧光素酶靶标验证系统验实ghr与igf2bp3为let-7b的靶标位点,let-7b的过表达会导致ghr、igf2bp3的mrna表达量明显下调,证明let-7b对ghr、igf2bp3基因具有负向调控作用,同时,发现let-7b通过介导ghr基因来影响jak-stat信号通路,从而调控骨骼肌的生长发育。还有许多报道表明,mirnas广泛的参与到鸡胚的生长发育、性成熟、神经细胞和肌肉细胞的增殖和分化和细胞迁移[29-33]等一系列生命活动中。
单核苷酸多态性(singlenucleotidepolymorphisms,snp)是指生命个体基因组序列中单个核苷酸发生的突变。snps在dna水平上出现的频率非常高,在所有突变形式中高达九成以上,但其中很多突变都集中在非编码区,其他很少的突变分布在基因的编码区。有证据显示,snp可以对rna、dna和蛋白水平对基因的表达及功能产生影响,是导致个体与群体之间的包括疾病发生,药物敏感与表型差异等的重要因素,其成熟体和前体序列上的snp有改变其形成过程甚至改变成熟体的靶标位点或改变与靶基因的结合强度,致使mirna的功能发生变化,从而致使动物性状的变异。每个mirna都有不止一个靶标位点,这就意味着位于mirna上的snp有着重要的意义,因此筛选mirna成熟体序列和前体序列上的snps,揭示其功能是很有研究意义的。当前查找筛选snp是用sanger测序或者pcr-rflp法来进行的,由于这些技术方法的效率较低,近些年来迅速发展的genechip技术和high-throughputsequencing能够在基因组水平上进行高效率的筛查突变位点。研究表明位于mirna种子区的snp更是稀缺,而存在于mirna前体区的snp数量较多被广泛应用于遗传学诊断、肿瘤易感性等领域。
mirna的基因多态可通过改变mirna的靶标位点和改变mrna的稳定性来影响转录前和转录后的调控,随后影响基因的表达以及表型的不同。这些多态位点可望作为分子标记用于育种中的辅助选择。zhao等在研究313只位于绵羊外显子2的rxrg基因突变及其与双胎性状的关系,通过pcr-sscp分析了rxrg基因第1外显子、第2外显子、第10外显子的基因多态性,结果表明,3种p2片段基因型与群体中的双胎性状显著相关(p<0.05)。蓝贤勇等通过45份内蒙古白绒山羊α-乳白蛋白(lalba)基因进行测序和分型,并与羊绒量、羊绒厚、羊绒长和体重性状进行关联分析。结果显示,其外显子3区域存在1个突变位点g.1897t>c。该位点不同基因型与产绒量存在显著相关(p<0.05),其tc基因型个体产绒量比tt基因型个体多产绒142.68g,高达26.21%(p<0.05)。lei等(2010)实验证明mir27a基因上的t/c突变与猪的每窝产仔数有关。huang采用pcr限制性片段长度多态性和微测序方法对187只鸭进行了snp基因分型。cc基因型和tt基因型的孵化率分别为79.59±3.40和76.35±1.77,显著高于ct基因型(65.77±2.07)(p<0.05)。
let-7家族是由reinhart等人在本世纪初发现的一类mirna。let-7家族成员分别位于几个不同的染色体上。研究发现let-7家族成员之一的let-7b靶向调节的基因在许多信号通路中的扮演重要角色,许多信号通路与细胞的增殖凋亡有关,因此有许多研究者研究let-7b与细胞增殖之间的关系。schμltz等研究发现let-7b抑制了黑色素瘤细胞的细胞周期进程和不依赖于锚定的生长。zhao等在研究中发现let-7b可以通过靶向干细胞调节因子tlx与细胞周期调节因子cyclind1来调节神经干细胞的增殖与分化;敲除则促进神经干细胞增殖,促进神经分化;而过量表达let-7b可降低神经干细胞增殖。陈楠在斑马鱼实验中发现,抑制低氧诱导因子1(hif-1a)会导致let-7b的表达下调,在zf4细胞中过表达let-7b会改变细胞周期过程,阻滞细胞周期中g1期到s期的过度,停止或减缓细胞的增殖;还通过体外实验鉴定了let-7b的靶基因foxh1也直接或间接的参与了细胞增殖过程。曲杰通过将mirlet-7b与anti-mirlet-7b转染至乳腺癌mcf-7细胞后进行划痕实验,结果显示let-7b能够显著抑制乳腺癌mcf-7细胞的迁移能力,而anti-mirlet-7b组能够显著促进乳腺癌mcf-7细胞的迁移,证明let-7b能够抑制乳腺癌mcf-7细胞的迁移能力。
通过位于鸡let-7b前体区基因序列中的snp位点与经济性状进行关联分析可得其与腿肌重,腿肌粗蛋白含量等重要性状显著或极显著相关,初步推测mirna前体区snp可能通过影响mirna的加工过程从而影响成熟体的表达量,进而影响一系列调控表达过程。
技术实现要素:
本发明的目的在于通过构建不同snp组合的单倍型重组质粒,转染至鸡原代成肌细胞,测定不同单倍型对let-7b成熟体表达量的影响,从而获得具有高let-7b成熟体表达量的单倍型鸡成肌细胞。
本发明所采取的技术方案是:一种具有高let-7b成熟体表达量的单倍型鸡成肌细胞的构建方法,其包括如下步骤:
1)单倍型表达载体克隆
a、根据let-7b前体区基因序列测序结果,设计引物对,目的片段包含g.270g>a、g.526g>a、g.678t>c和g.710g>c4个突变位点,同时在设计的引物对的5'端加上酶切为点和保护碱基后,进行单倍型基因pcr扩增,得pcr产物;
b、pcr产物进行回收、纯化,获得纯化的含有目的片段的dna液,备用;
c、采用pcdna3.1菌种进行菌液扩繁,抽提pcdna3.1环状质粒并获取纯化的dna样品;
d、用内切酶分别对步骤b所得的含有目的片段的dna液和步骤c所得的dna样品进行双酶切处理,经纯化双酶切产物后获得目的基因片段和pcdna3.1(+)质粒载体;
e、使用t4dnaligase将步骤的d所得的目的基因片段分别与pcdna3.1(+)质粒载体连接,将连接产物进行转化,再进行摇菌扩繁处理,获得构建成功的5种单倍型重组质粒;
2)鸡原代成肌细胞培养
利用孵化11d的鸡种蛋分离出成肌细胞,纯化处理,备用;
3)细胞转染
采用
本发明的有益效果是:本发明构建出不同单倍型过表达载体,并转染至鸡原代成肌细胞以测定不同单倍型对let-7b成熟体表达量的影响,从而获得具有高let-7b成熟体表达量的单倍型鸡成肌细胞,推断出其表达量差异的原因是因为let-7b前体区上不同的snp影响let-7b前体加工过程,从而导致let-7b成熟体表达量差异。
附图说明
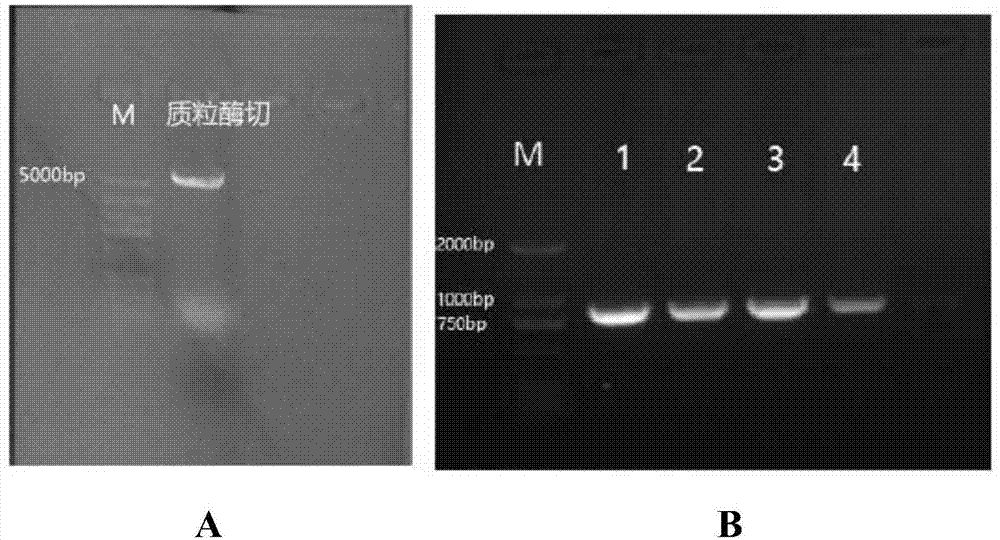
图1为2.6中的双酶切结果(a为pcdna3.1双酶切电泳图;b为不同单倍型双酶切电泳图);
图2为2.11中的部分菌液鉴定电泳图;
图3为2.11中菌液测序结果(a为菌液测序结果中上游酶切位点;b为菌液测序结果中下游酶切位点);
图4为2.11中引入的4个突变位点情况(a为菌液测序中g.270a>g突变位点;b为菌液测序中g.526a>g突变位点;c为菌液测序中g.678t>c突变位点;d为菌液测序中g.710g>c突变位点);
图5为3.2中的鸡原代成肌免疫细胞荧光(×40)的拍照结果(a为dapt染色;b为desmin荧光蛋白染色);
图6为3.9中不同单倍型转染let-7b相对表达量(其中纵轴1代表agcc单倍型;2代表aacc单倍型;3代表gatc单倍型;4代表gacg单倍型;5代表gacc单倍型;6代表pcdna3.1空载体)。
具体实施方式
下面结合实施例对本发明进行具体描述,以便于所属技术领域的人员对本发明的理解。有必要在此特别指出的是,实施例只是用于对本发明做进一步说明,不能理解为对本发明保护范围的限制,所属领域技术熟练人员,根据上述发明内容对本发明作出的非本质性的改进和调整,应仍属于本发明的保护范围。同时下述所提及的原料未详细说明的,均为市售产品;未详细提及的工艺步骤或制备方法为均为本领域技术人员所知晓的工艺步骤或制备方法。
一、实验材料
1.1实验动物
分离成肌细胞的种蛋购自华南农业大学种鸡场。
1.2主要试剂及配制溶液
dna纯化回收试剂盒购自omeg公司;trizol与rna固相清除剂购自invitrogen公司;质粒抽取试剂盒购自omeg公司;sybrgreenpcrmastermix定量试剂盒购自日本toyobo公司;mirnaqrt-pcr反转录试剂盒购自广州市锐博生物科技有限公司;1640培养基、胎牛血清、anti-desmin、
lb液体培养基:4gtryptone,2gyeastextract,4gnacl,双蒸水(ddh2o)定容至400ml,高压灭菌后4℃保存。
la液体培养基:4℃保存的400mllb液体培养基加入氨苄青霉素400μl,即为la液体培养基。
含氨苄青霉素固体la培养基:4gtryptone,2gyeastextract,4gnacl,10g琼脂粉,双蒸水(ddh2o)定容至400ml,高压灭菌。待其冷却至不烫手左右时加入氨苄青霉素,充分混匀后分装至灭菌培养皿内,待其凝固后4℃保存。
氨苄青霉素贮存液:用双蒸水将氨苄青霉素配成100mg/ml,用0.22mm滤膜过滤除菌,-20℃贮存,工作浓度为100mg/ml。
二、不同单倍型表达载体克隆
2.1引物设计
根据let-7b前体区基因序列测序结果,利用primerpremier5.0软件设计引物对,要求目的片段包含g.270g>a、g.526g>a、g.678t>c和g.710g>c4个突变位点,同时在设计的引物的5'端加上合适的酶切位点和保护碱基,引物对信息如表1所示,其中加粗部分为保护碱基序列,标有下划线部分为酶切位点(ecorl和xbal)。
表1引物对信息
2.2单倍型基因扩增
pcr扩增反应体系按照下表2来配制,pcr反应程序为:94℃预变性2min,40个循环(98℃变性10s,54℃退20s,72℃延伸2min30s),72℃后延伸10min,4℃保存。pcr产物用1.5%的琼脂糖凝胶电泳检查,检测合格的产物进行下一步实验。
表2let-7b前体区pcr反应体系
2.3pcr产物的回收和纯化
将电泳检测合格的pcr产物,进行切胶回收、纯化。方法参照hipuregelpurednaminikit(magen)琼脂糖凝胶dna回收试剂盒说明书来操作,获得纯化的含有目的片段的dna液,备用。
2.4菌液的扩繁
pcdna3.1菌种为本实验室保存的菌种。移液枪吸取200μl菌种接种到20mlla液体培养基中,200rpm37℃摇床震摇8h后取出抽提载体质粒。
2.5pcdna3.1环状质粒抽提
(1)取出菌液室温3000-5000rpm,离心10min,弃上清;
(2)往细胞沉淀中加500μl带有rnasea的buffere1,高速涡旋震荡15s重悬菌液,移至新的2ml离心管;
(3)往细胞悬液中加500μl溶液buffere2,轻轻混匀颠倒离心管8~10次,切勿震荡,混合液变得粘稠透亮。室温静置2min;
(4)加入500μl预冷的buffere3,立即轻轻上下颠倒离心管10~15次混匀,产生白色沉淀,切勿震荡(此步骤为质粒产量的关键步骤),13000×g离心10min;
(5)将hipureefminicolumn套在收集管上,转移750μl上清液到柱子中,加0.1倍体积的etrsolution,颠倒混匀数次,冰育10min(期间颠倒数次),液体逐渐澄清;
(6)接着水浴5min,此时管中液体又变浑浊。12000×g离心3min;
(7)转移上清液到另一干净1.5ml管中,加0.5倍体积的无水乙醇,轻轻颠倒6~7次混匀,室温放置1~2min;
(8)转移上一步混合液于干净的hibindtmdnaminicolumus吸附柱中,10000×g离心1min;
(9)弃滤液,继续转移(7)中液体,直到全部过柱;
(10)向吸附柱中加500μlbufferhb,10000×g离心1min;
(11)向吸附柱中加700μldnawashingbuffer,10000×g离心1min;
(12)重复(11)一次;
(13)弃滤液,空柱13000×g离心2min;
(14)将吸附柱放于新的1.5ml离心管中,加45μl的bufferte溶解质粒;
(15)获取纯化的dna样品,保存于-20℃。
2.6双酶切
用内切酶ecorl和xbal分别对2.3所得的含有目的片段的dna液和2.5所得的dna样品进行双酶切处理,酶切体系如下表3所示,在37℃培养箱酶切2h,双酶切结果如图1a和图1b所示,通过将加了酶切位点的不同单倍型的扩增产物和pcdna3.1质粒进行双酶切实验得出图1a和1b的电泳图,产物大小符合预期产物长度,可用于后续试验。
表3双酶切体系
2.7纯化双酶切产物
采用omega的dna纯化回收试剂盒回收pcr产物,pcr产物回收方法如下:
(1)瞬时离心pcr产物,用移液枪测量产物体积,将其移至1.5/2.0ml的离心管中;
(2)加入等体积的buffergdp,颠倒或涡旋将其混匀;
(3)将hipuredna柱子套在收集管中,将混合液移至柱子中1000×g离心30~60s;
(4)弃掉滤液,把柱子重新套回收集管中,加入600μlbufferdw2(已用无水酒精稀释过的,dw2:无水酒精=1:4)到柱子中,12000rpm离心30~60s;
(5)弃掉滤液,把柱子重新套回收集管中,加入600μlbufferdw2(用无水酒精稀释过的,dw2:无水酒精=1:4)到柱子中,12000rpm离心30~60s;
(6)弃掉滤液,将柱子重新装回柱子中,12000rpm离心2min;
(7)把柱子装在新的1.5ml离心管上,加入45μl左右55℃预热的elutionbuffer到柱子膜中央,室温静置2min,12000rpm离心1min以洗脱出dna;
(8)电泳检测纯化产物双酶切产物,于-20℃保存备用。
2.8连接
使用t4dnaligase将纯化后的目的基因片段分别与pcdna3.1(+)质粒载体连接,酶链仪16℃连接过夜,连接体系如下表4所示。
表4t4dna连接酶连接体系
2.9转化
对连接产物进行转化,首先在超净台中配制转化体系,体系组分如下表5所示。
表5连接产物转化体系
转化方法为:将转化体系冰盒中冰浴25min,之后酶链仪中42℃热激1min,在放置冰盒中冰浴2min随后在超净台中,将转化体系与500μl的不含氨苄抗性的lb混匀于37℃200rpm摇床震摇1h,结束后3000rpm离心4min,弃掉300μl的上清,将剩下的混合物轻轻重悬,在超净台中涂布于含有氨苄青霉素的la平板37℃恒温培养箱过夜培养。
2.10摇菌
等到la板长出菌株,在超净台中挑取单菌落至加有800μlla液体培养基的1.5ml离心管中,将离心管置于37℃200rpm摇床震摇4h扩繁。
2.11阳性菌落pcr及测序鉴定
将扩繁好的菌液进行菌液pcr以及测序,目的是检测目的片段是否与pcdna3.1载体连接成功。菌液pcr的体系如下表6所示,pcr反应程序为:94℃预变性5min,38个循环(98℃变性30s,56℃退火30s,72℃延伸30s),72℃后延伸7min,4℃保存。
表6菌液pcr体系
将菌液pcr产物进行1.5%琼脂糖凝胶电泳检测,使用通用引物t7对其进行pcr菌液鉴定,鉴定结果如附图2所示,电泳图中的单条带即为构建成功的单倍型表达载体,其含目的片段的载体条带为1000bp左右,将条带大小符合的菌样送生工生物科技有限公司测序,其测序结果如附图3a、3b,表明加了酶切位点进行pcr扩增,成功将酶切位点引入目的序列,根据附图3a中酶切位点两翼序列比对,左边为pcdna3.1载体序列,右边为不同单倍型目的序列,根据附图3b中酶切位点左边为不同单倍型目的序列,右边为pcdna3.1载体序列,表明两端序列连接成功。引入的4个突变位点情况分别如附图4a、4b、4c和4d所示,表明载体构建成功。用seq-man软件打开菌液测序结果获得所有单倍型,共5种,如下表7所示。将测序正确的菌样进行扩繁保种,以备后续试验使用。
表7let-7b前体单倍型
三、原代成肌细胞的培养
3.1成肌细胞的分离和纯化
(1)将孵化11d的种蛋从孵化箱取出,用75%擦拭蛋壳消毒。用镊子大头轻轻敲破种蛋气室,用镊子小心取出鸡胚;
(2)用手术刀轻轻刮掉腿部皮肤,再分离鸡胚腿部,置于含20%胎牛血清的1640(原代成肌细胞培养液)培养基中;
(3)用镊子分离弃去皮肤和骨头,置于1.5μl的离心管中,3~4个腿肌放置一个离心管,每管加入500μl培养基,用剪刀将腿肌组织剪至1ml枪头可以吸起来为止;
(4)将剪碎的肌肉组织用移液器转移到50ml离心管中,涡旋震荡1min,再静置2min,待肌肉组织慢慢沉到底部。转移上清液通过200目过滤筛过滤至50ml离心管;
(5)添加7ml原代成肌细胞培养液重悬肌肉组织,涡旋振荡1min,重复此步骤3~4次;
(6)对过滤收集到的滤液进行1050rpm离心4min,弃上清;
(7)加入适量培养基重悬获得的细胞,转移至新的培养皿中,放置37℃,5%co2细胞培养箱中培养。通过两次差速贴壁法得到较为纯化的成肌细胞。
3.2原代成肌细胞的免疫荧光鉴定
desmin是一种成肌细胞特异表达蛋白,与成肌细胞特异结合后呈现绿色荧光。dapi是一种核酸染料,能够穿过活细胞的细胞膜进入细胞内与细胞核中的脱氧核糖核酸相结合,其在荧光显微镜下的细胞核呈现为蓝色。通过免疫荧光染色鉴定成肌细胞,分离获得的细胞大部分为成肌细胞,可以用于后续实验。鸡胚成肌细胞的免疫荧光步骤如下:
(1)待到24孔板中原代成肌细胞密度达到90%时,用pbs100μl每孔清洗细胞3次,每次5min;
(2)弃掉pbs,每孔加250μl固定液(4%冷的多聚甲醛),室温静置20min;
(3)加适量的pbs,室温下每5min用pbs洗一次,洗3次;
(4)每孔加1ml的0.1%tritonx-100破膜液,室温下破膜15min,弃掉破膜液,室温下每5min用pbs洗一次,洗3次;
(5)加bsa封闭液,200μl/孔,室温下封闭30min;
(6)弃去封闭液,不用pbs洗,加desmin4℃过夜;
(7)翌日,将板从4℃冰箱中取出,每5min用pbs洗一次,洗3次,然后每孔加1:50稀释的异硫氰酸荧光素(fitc),避光孵育1h,每5min用pbs洗一次,洗3次;
(8)dapi染色液200μl/孔染核5min。染核后每5min用pbs洗一次,洗3次;
(9)在载玻片上滴上一滴甘油,荧光倒置显微镜下观察,并拍照,拍照结果如附图5a、5b所示。
3.3细胞铺板
(1)待培养皿中的原代细胞密度达到100%时,在无菌操作台弃掉培养基,每皿用1mlpbs洗2次,去除血清和死细胞,随后用移液枪弃掉;
(2)1ml胰酶每皿消化30s后,在倒置显微镜下观察,待细胞收缩变圆,轻轻吹打使细胞脱落,培养基终止消化,用200目纱网过滤(筛网下接50ml离心管)后室温1050rpm离心5min;
(3)弃掉上清,加入新鲜含20%胎牛血清的1640培养基振荡器重旋细胞,用移液器将细胞悬液均匀打入24孔板,每孔定容至500μl。放入37℃5%co2培养箱中培养。
3.4细胞转染
对原代成肌细胞转染5个单倍型重组质粒和一个pcdna3.1空载体质粒,探究不同单倍型在成肌细胞中对let-7b表达量的影响,细胞转染的步骤方法按
(1)将原代成肌细胞接种到24孔板中,待细胞密度达到75%左右时即可进行转染实验,每个单倍型转染有4个重复;
(2)opti-mem每孔50μl/孔,每个重组质粒添加600ng/孔,p30002μl/孔;
(3)用50μl/孔opti-me来稀释
(4)将(2)和(3)混合后室温孵育5min;
(5)每孔细胞添加50μl混合液,轻摇细胞培养板数次,混匀,将24孔板放回37℃细胞培养箱中培养;
(6)转染6h后弃掉培养基,pbs洗一遍,加入新鲜的含20%血清的1640培养基;
(7)培养48h后拿出,进行下一步试验。
3.5rna抽提
(1)将24孔板内培养基弃掉,加入1mltrizol溶液吹打几下,室温静置5min;
(2)12000r/m,4℃离心15min,将上清液转移至新的1.5mlep管;
(3)加入200μl氯仿,剧烈振荡混匀15s,室温静置5min;
(4)4℃下,12000r/m离心15min,移上清液于新的ep管中;
(5)加入与上清液等体积约500ml的异丙醇,-80℃放置30min;
(6)4℃下12000r/m离心10min,弃去上清液,加入75%乙醇1ml洗涤(适度振荡混匀)沉淀;
(7)4℃下7500r/m离心5min,弃上清保留沉淀,室温下自然干燥5min;
(8)将干燥的沉淀溶解于适量(3~50μl)的depc处理水中,得到rna溶液。
3.6总rna浓度测定
总rna完整性检测可通过普通琼脂糖凝胶电泳检测,过程如下:
(1)制备1%琼脂糖凝胶:称取0.4g琼脂粉,加40ml1×tae,放在微波炉加热1min,待冷却到60℃左右,加入1.5μled染色混合晃匀,倒入凝胶盒中,除掉气泡,插上合适的梳子。室温冷却30min,待凝胶凝固后,拔掉梳子,即可使用;
(2)点样:吸取3μl总rna与2μl6×loadingbuffer用移液枪吹打之后加入点样孔中,以2kdnamarker作为对照,电泳120v,12min,(电泳开始之前换成新配的电泳液);
(3)拍照:凝胶电泳结束后,用凝胶成像系统观察并拍照,标准的总rna共有三条电泳带,分别为5srrna,18srrna和28srrna,如果三条带完整说明本次总rna抽提效果较好,可以用于后续试验;
(4)总rna纯度检测:用紫外分光光度计测量所抽提总rna浓度,每个样品都要重复测量至少3次,平均值作为其最终浓度。依照该平均值,用depc水将每个rna样品稀释至1μg/μl,置于-80℃超低温冰箱保存备用。
3.7总rna反转录
(1)在rnasefreepcr管中加入如下表8所示的试剂;
(2)将上述发转录体系42℃加热60min;
(3)72℃加热10min后,将反转录产物cdna置于-20℃保存备用。
表8总rna反转录反应体系
3.8qpcr
用于qpcr检测样品中mirna含量的反应体系如下表9所示,使用bio-radcfx96实时荧光定量pcr仪进行。反应程序为:预变性95℃30s,变性95℃5s,退火60℃30s,40个循环。在40个循环跑完之后马上分析其溶解曲线,检测温度为65~95℃,升温速率是0.5℃/次,恒温时间为5s/次。
表9let-7b的qpcr反应体系
3.9qpcr的数据分析处理
采用比较ct值法进行基因相对表达量分析。△ct=ct(目的基因)-ct(内参基因);△△ct=△ct(实验组)-△ct(对照组),相对表达量=2-△△ct。
根据荧光定量数据相对表达量=2-△△ct的方法计算出5个单倍型和一个pcdna3.1空载体质粒过表达至鸡原代成肌细胞48h后let-7b成熟体的表达量,结果如附图6所示,其中agcc单倍型转染后let-7b成熟序列相对表达量为2.362,aacc单倍型为3.188,gatc单倍型为3.649,gacg单倍型为3.052,gacc单倍型为2.977,而pcdna3.1空载体只有1.054。
上述实施例为本发明的优选实施例,凡与本发明类似的工艺及所作的等效变化,均应属于本发明的保护范畴。
- 还没有人留言评论。精彩留言会获得点赞!