一类新型可光控释放的核磁造影脂质及其制备方法与流程
本发明涉及可光控释放的核磁造影脂质体及其合成方法,其属于表面活性剂合成化学领域。本发明涉及的t1弛豫时间增强光控释放系列化合物是一类可自组装形成囊泡、可光解、可追踪磁共振信号的纳米药物载体,其中的烷基碳原子总数为6~36。
背景技术:
随着生物医药材料科学的发展,新型可控药物释放系统己成为药学领域的重要研究方向。可控药物传输系统有利于提高药效,降低药物的毒副作用,在疾病治疗和医疗保健等方面应用前景广阔。而降低药物毒性和提高药物效率,一直是药物领域的重大科学问题和关键瓶颈技术。基于我国各大医院都有核磁共振仪及现有纳米药物载体存在的功能单一的问题,本专利提出精准治疗的多功能纳米药物载体。
目前国内外纳米药物载体的研究主要集中在可视(荧光可视和磁性可视)方向,因生物自体放光等限制,磁性可视有更广阔的应用前景,如tatjana等人将1,10-菲咯啉通过酰胺键连接到二乙烯三胺五乙酸(dtpa)分子中间的羧基上,得到的配体络合上钆(iii),三个钆(iii)配合物可以围绕铁(ii)离子自组装。这种钆(iii)-铁(ii)络合物比母体gd-dtpa络合物有更高的弛豫率。在20mhz,310k下其弛豫率为9.5±0.3s-1·mm-1,高于gd-dtpa络合物(3.9s-1·mm-1)。又如jung等报告了dota的2,2'-二氨基联苯衍生物及其gd型络合物,其水中的t1弛豫效率达到7.3mm-1s-1,比医用gd-dota多出一倍。而另一方面,为了降低毒性,增加药效,部分国内外研究者开始研究可控释放型脂质(ph控释、光控释、有机/无机复合物控释),其中光响应药物载体是脂质体中的一个新兴类型,其具有使用条件简单且副作用小等优点,光降解化合物在受光激发后,分子内化学键发生断裂,有效释放被载体保护的药物分子,以完成药物的控释。如kang等人合成了以邻硝基苄酯键为结点连接亲水基和亲油基的聚合物,在紫外光照射下破坏亲水亲油平衡,实现药物的快速释放。han等人合成了以邻硝基双苄酯为重复单元的聚合物,使其在紫外光照射下主链迅速断裂,从而引起药物的快速释放。
本专利提供一种可视和可控释放多功能的药物载体,将两个功能结合到一起,实现囊泡在体内运输的可追踪能力以及可光控释放能力,以实现精准治疗。本专利从分子设计出发,将双长链、靶向性基团和光照快速释放基团引入核磁造影剂母体结构中,合成具有可视和可控释放功能的精准治疗的纳米载体。所设计的两亲性分子结构既是造影剂,实时精确可视监控,又可经光照后快速释放药物,是可视和光控释药的多功能释药体系。同时,两亲性分子自组装形成囊泡作为造影剂也尚未见文献报道。
技术实现要素:
本发明的目的在于提出一类含有邻硝基苄酯光降解基团的t1弛豫时间增强脂质的结构及其制备方法,其特征在于,分子结构中含有核磁造影基团(有且不限于dtpa、乙二胺四乙酸(edta)、dtpa-bsa、hp-do3a或dota中的一种)、4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸结构(分子基团中至少带有一个或以上的羧基与一个或以上的氨基的小分子化合物)、光裂解基团(邻硝基苄酯键)、以及对称或不对称长链仲胺或伯胺。该结构制备方法可分成四个部分:多羧基化合物的单酐合成(如二乙烯三胺五乙酸单酐的合成);对脂肪酰胺邻硝基苄酯琥珀酸丙烯酯的合成;疏水端与亲水端的连接反应(如最终产物二乙烯三胺五乙酸对脂肪酰胺邻硝基苄酯琥珀酸丙烯酯的合成);多羧基产物的金属螯合与提纯。
本发明提出了一种新型的可视可控囊泡的结构以及合成路线,向多羧基核磁造影基团上引入带有光控基团的双长碳链结构,赋予其形成囊泡的能力,当mri检测到病变组织造影效果明显增强时,即为囊泡运行到病变区域。此时,施加紫外照射手段,刺激病变区域,邻硝基苄酯结构破坏、囊泡结构破坏,达到释放药物的作用,从而提高药效降低毒性。因此本专利提出一种具有实时监控及控制释放的新型可视-光控释放纳米药物载体,可作为一种潜在的治疗手段。
本发明提供一种含有邻硝基苄酯光降解基团的t1弛豫时间增强脂质,其结构如通式ⅰ,
式中,cnh2n+1,n=6,8,10,12,14,16,18;cmh2m+1,m=0,6,8,10,12,14,16,18;r1选自下述基团中的一种,
本发明提供上述的一种含有邻硝基苄酯光降解基团的t1弛豫时间增强脂质的制备方法,其特征在于包括以下步骤:
(1)脱水反应:将多羧基化合物、无水乙酸酐和无水吡啶加入到单口烧瓶中,氮气保护下,加热至回流,反应12~24h后,抽滤,洗滤饼至无色,50~90℃真空干燥,得到多羧基二酐;所述多羟基化合物选自二乙烯三胺五乙酸、乙二胺四乙酸、dtpa-bsa、hp-do3a或dota中的一种;
(2)单酐反应:将(1)中得到的多羧基二酐溶于无水溶剂中,升温至80~100℃,搅拌使其完全溶解。取等摩尔量的去离子水,缓慢滴加到反应溶液中,于80~100℃下,搅拌反应2~5h。
(3)酰化反应:将对羧基邻硝基苄溴与酰化试剂投入单口烧瓶中,在氮气保护下,加热回流,反应6~8h后,得到黄色油状液体,旋蒸后柱色谱分离,得到对羧酰氯邻硝基苄溴。
(4)酰胺化反应:将仲胺或伯胺加入(3)得到的对羧酰氯邻硝基苄溴的有机溶液中,滴加适量的碱,在氮气保护下,加热回流,反应16~24h后,得到橘黄色油状液体,冷却到室温,柱色谱分离,得到带疏水长碳链的对脂肪酰胺基邻硝基溴苄。
(5)酯化反应:将4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸溶于有机溶剂,室温下滴加催化剂,室温搅拌30~60min。室温下加入(4)得到的对脂肪酰胺基邻硝基溴苄,室温搅拌12-24h,反应完全后,旋蒸溶剂,柱色谱分离,得到带疏水长碳链的对脂肪酰胺基邻硝基苄酯衍生物
(6)去保护基反应:将(5)中得到的反应产物溶于三氟乙酸,室温搅拌1~4h,多次旋蒸除去溶剂,真空干燥箱中干燥,柱色谱分离,得到具有光裂解能力的1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)2-(氨基)琥珀酸-4-丙烯酯。
(7)将干燥好的(6)得到的产物溶于有机溶剂中,于80~100℃缓慢滴加到上述的单酐中,搅拌反应24h。反应完全后,除去多余溶剂,柱色谱提纯,得到1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)-2-(多羧酸基酰胺)琥珀酸-4-丙烯酯。
(8)螯合反应:取上部纯品溶于极性溶液中。加入适量金属螯合剂,搅拌2~5h,析出淡黄色絮状固体,过滤,干燥,柱色谱分离,得到金属螯合的1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)-2-(多羧酸基酰胺)琥珀酸-4-丙烯酯。
本发明提供一种t1弛豫时间增强可光解脂质,以r1为二乙烯三胺五乙酸为例,其结构如通式ⅱ,
式中,cnh2n+1,n=6,8,10,12,14,16,18;cmh2m+1,m=0,6,8,10,12,14,16,18。
本发明t1弛豫时间增强可光解脂质式ⅱ的合成方法,包括以下步骤:
(1)脱水反应:将二乙烯三胺五乙酸、无水乙酸酐和无水吡啶加入到单口烧瓶中,氮气保护下,加热至回流,反应24h后,抽滤,洗滤饼至无色,80~100℃真空干燥。
(2)单酐反应:将二乙烯三胺五乙酸双酐溶于无水溶剂中,升温至80℃,搅拌使其完全溶解。取等摩尔量的去离子水,缓慢滴加到反应溶液中。于80~100℃下,搅拌反应2~5h。
(3)酰化反应:将对羧基邻硝基苄溴与酰化试剂投入单口烧瓶中,在氮气保护下,加热回流,反应16~24h后,得到黄色油状液体,旋蒸后,柱色谱分离。
(4)酰胺化反应:将长链仲胺或伯胺加入对羧酰氯邻硝基苄溴的有机溶液中,滴加适量的碱,在氮气保护下,加热回流,反应16~24h后,得到橘黄色油状液体,冷却到室温,柱色谱分离,得到产物。
(5)酯化反应:将4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸溶于有机溶剂,室温下滴加催化剂,搅拌30~60min。室温下加入对酰胺基邻硝基苄溴,室温搅拌12-24h,反应完全后,旋蒸溶剂,柱色谱分离。
(6)去保护基反应:将上步反应产物冰水浴条件下溶于三氟乙酸,室温搅拌1~4h,多次旋蒸除去溶剂,真空干燥箱中干燥,柱色谱分离。
(7)将干燥好的1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)2-(叔丁氧基羰基)琥珀酸-4-丙烯酯溶于有机溶剂中,80~100℃下缓慢滴加到上述二乙烯三胺五乙酸单酐液中,保持温度搅拌反应24h。反应完全后,除去多余溶剂,柱色谱提纯。
(8)螯合反应:取纯品1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯于溶剂中。加入适量金属螯合剂,室温搅拌2~5h,析出淡黄色絮状固体,过滤,干燥,柱色谱分离。
以上八步反应中,第一步的投料量为二乙烯三胺五乙酸:吡啶:乙酸酐摩尔比1:4~6:8~10。第二步的投料量为二乙烯三胺五乙酸双酐:去离子水摩尔比为1:1。第三步的投料量为对羧基邻硝基苄溴:酰化试剂摩尔比为1:12~20。第四步的投料量为对酰氯邻硝基苄溴:仲胺或伯胺:碱的摩尔比为1:1.5:2~3。第五步的投料比为4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸:催化剂:对脂肪酰胺基邻硝基苄溴摩尔比为1:1.2:1。第六步的投料量为含有的boc基团:三氟乙酸的摩尔比为1:20。第七步的投料量为1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)2-(叔丁氧基羰基)琥珀酸-4-丙烯酯:二乙烯三胺五乙酸单酐摩尔比为1~2:1。第八步的投料量为1-(4-(长链烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯:金属螯合剂摩尔比为1:10。
进一步地,在上述技术方案中,步骤(2)(7)中,所述的有机溶剂为n,n-二甲基甲酰胺、二甲基亚砜、乙腈、甲酰胺、六甲基磷酰胺、四氢呋喃中的一种。
进一步地,在上述技术方案中,步骤(3)中,所述的酰化试剂为乙酸酐、邻苯二甲酸酐、乙酰氯、二氯亚砜、甲基磺酰氯、硫酸二甲酯中的一种。
进一步地,在上述技术方案中,步骤(4)中,所述的碱溶液为三甲胺、三乙胺、三乙醇胺、吡啶、n-甲基吡啶、氢氧化钠、氢氧化钾、碳酸钾或碳酸钠中的一种。
进一步地,在上述技术方案中,步骤(5)中,催化剂为dcc,edc,hbtu,dbu,tbtu,nhs中的一种。
进一步地,在上述技术方案中,步骤(8)中,螯合反应中所述的金属络合剂为氯化钆、氧化钆、氧化铁、氯化铁中的一种。
进一步地,在上述技术方案中,步骤(8)中,螯合化反应中所述溶剂选水、乙腈、甲醇与氯仿混合液、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜与水中的一种。
进一步地,在上述技术方案中,本申请化合物,含有能核磁造影基团,有且不限于dtpa、edta、dtpa-bsa、hp-do3a和dota的一种。
进一步地,在上述技术方案中,本申请化合物,疏水端为对称或不对称长链仲胺或伯胺,cnh2n+1,n=6,8,10,12,14,16,18;cmh2m+1,m=0,6,8,10,12,14,16,18。
本发明制备的上述化合物可形成光控释放、体内核磁追踪型多功能囊泡,在纳米药物载体和可光解的核磁造影剂中应用。
发明有益效果
本发明钆-1-(4-(脂肪烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-单酰胺五乙酸三胺二乙烯类化合物结构新颖,形成的囊泡可体内追踪药物运输情况,当囊泡运输到病变区域后,即光解控释药物释放,是多功能精准治疗型囊泡。
(1)本发明利用4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸的多羧酸以及氨基的结构特性,巧妙地避免了如二乙烯三胺五乙酸酐类化合物的对称双酐反应活性高,难以选择反应的缺点,实现了选择性的反应,得到两端不对称的两亲性分子。
(2)本发明将二乙烯三胺五乙酸类化合物的金属络合基团加到长链疏水端上,按照sbm理论,通过增加分子的分子量来提高分子的弛豫时间,赋予脂质体t1弛豫时间增强效应,以便其形成的囊泡可以在体内被追踪到。
(3)本发明利用有机碱的高反应催化活性,苄溴出发与羧酸反应成酯,单相反应,提供了一种新的酯化反应路径。
(4)本发明的脂质体所形成的囊泡,经不同时间的紫外光照(365nm),空白囊泡的粒径不断增加,由150nm增加至1000nm,而载药囊泡经不同的光照时间,其粒径先增大后急剧减小,由260nm先增加至600nm,后急剧减小到100nm,说明可光控释放,因为分子光裂解后生成更多的疏水基团,因疏水性而与未裂解的两性分子自组装形成更大的囊泡。空白囊泡随不同光照时间的动态光散射如图1所示,空白囊泡粒径形貌光照前与光照120min后的变化如图2所示。载药囊泡随不同光照时间的动态光散射如图3所示,载药囊泡粒径形貌光照前与光照120min后的变化如图4所示。
(5)本发明分子的体外弛豫时间及核磁共振成像性能是现有的gd-dtpa核磁造影剂的4倍,同摩尔浓度下,本发明的脂质溶液成像更加明亮,是一种优秀的t1增强造影剂,同时也是一种潜在的可视型脂质。以所合成的钆-双十二-二乙烯三胺五乙酸脂质(gd-dtpa-obn12)与临床所使用的gd-dtpa造影剂作对比,弛豫时间对比图如图5所示,体外造影成像图如图6所示。
附图说明
图1为实施例1制备的gd-dtpa-obn12所形成的的空白囊泡随不同光照时间的动态光散射变化图;
图2为实施例1制备的gd-dtpa-obn12所形成的空白囊泡粒径形貌光照前与光照120min后的变化图;
图3为实施例1制备的gd-dtpa-obn12与阿霉素所形成的的载药囊泡随不同光照时间的动态光散射变化图;
图4为实施例1制备的gd-dtpa-obn12与阿霉素所形成的的载药囊泡粒径形貌光照前与光照120min后的变化图;
图5为实施例1制备的gd-dtpa-obn12与产品级gd-dtpa的弛豫时间对比图;
图6为实施例1制备的gd-dtpa-obn12与产品级gd-dtpa体外造影成像对比图;
图7为实施例1制备的二乙烯三胺五乙酸双酐(dtpaa)的1hnmr图;
图8为实施例1制备的4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺的1hnmr图;
图9为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-烯丙酯的质谱图;
图10为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸4-烯丙酯的1hnmr图;
图11为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-丙烯酯的质谱图;
图12为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-丙烯酯的1hnmr图;
图13为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯的质谱图;
图14为实施例1制备的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯的1hnmr图;
图15为实施例1制备的螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-单酰胺五乙酸三胺二乙烯的质谱图;
图16为实施例1制备的螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-单酰胺五乙酸三胺二乙烯的液相色谱图;
图17为实施例1制备的gd-dtpa-obn12与dtpa-obn12的红外光谱图;
具体实施方式
下面结合实施例对本发明的方法做进一步说明,但并不是对本发明的限定。
实施例1
(1)二乙烯三胺五乙酸双酐的合成(dtpaa)
将2gdtpa,2.5ml无水乙酸酐,3.5ml无水吡啶加入到50ml单口烧瓶中,氮气保护下,65℃加热回流,反应24h后,得到深棕色悬浮液,抽滤,滤饼用无水乙酸酐和无水乙醚交替洗至无色,80℃真空干燥12h,得到1.56g白色固体,熔程为178℃~181℃,产率85.9%。
(2)二乙烯三胺五乙酸单酐的合成
称取3.57gdtpaa加入到50ml三口烧瓶中,加入20ml无水dmf,升温至80℃,搅拌使其完全溶解。精确称取等摩尔量的去离子水0.18g于10ml无水dmf中稀释,缓慢滴加到反应溶液中。于90℃下,搅拌反应2.5h,冷却至室温,保存备用。
(3)对羧酰氯邻硝基苄溴的合成
将2.80g对羧基邻硝基苄溴与13g甲磺酰氯投入100ml单口烧瓶中,在氮气保护下,50℃加热回流,反应6h后,得到黄色油状液体,冷却到室温,分别添加无水氯仿30ml,三次旋蒸抽滤,得到淡黄色油状液体,80℃真空干燥12h备用,得到2.98g油状液体,直接用于下一步反应。tlc(二氯甲烷:甲醇=10:1v/v),rf=0.23。
(4)4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺
将3.80g双十二仲胺,投入干燥好的对羧酰氯邻硝基苄溴中,向100ml单口烧瓶中加入30ml乙腈,滴加无水三乙胺1.4g,在氮气保护下,加热回流,tlc检测监测反应进程,反应16h后,得到橘黄色油状液体,冷却到室温,收集有机相,用无水硫酸钠干燥2h。过滤有机相后,旋干液体,得到橘红色油状液体,于30℃真空干燥箱中干燥12h。柱色谱分离(二氯甲烷:甲醇=10:1v/v)rf=0.84,收集产品,旋蒸干燥,得到产物4.81g,产率81.57%。
(5)1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸4-烯丙酯的合成
向100ml单口烧瓶中加入干燥好的40ml四氢呋喃,投入2.73g4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸,室温下滴加1.83gdbu,升温至56℃,将4.81g4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺溶于10ml四氢呋喃,向圆底烧瓶中缓慢滴加,滴加结束后回流反应14h。旋干有机溶剂,收集有机相,无水硫酸钠干燥后过滤有机相,旋干液体,得到红棕色的粘稠油状液体,室温下真空干燥12h。柱色谱分离(二氯甲烷:甲醇=10:1v/v)rf=0.70,旋蒸,干燥后得到产物5.03g,产率78.40%。
(6)1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-烯丙酯的合成
取提纯后的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)2-(氨基)琥珀酸4-烯丙酯5.03g于100ml圆底单口烧瓶中,滴加无水三氟乙酸18g后,搅拌4h,多次旋蒸除去溶剂,30℃真空干燥箱中干燥12h。柱色谱分离(二氯甲烷:甲醇=10:1v/v)rf=0.32,旋蒸,干燥后得到产物3.62g,产率82.64%。
(7)1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(dtpa-obn12)的合成
将干燥好的3.62g的1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)2-(叔丁氧基羰基)琥珀酸-4-丙烯酯溶于10ml干燥的dmso中,80℃下缓慢滴加到上述二乙烯三胺五乙酸单酐液中,保持冰水浴搅拌反应24h。tlc追踪反应,反应完全后,将dmso旋干,30℃真空干燥。两次柱色谱提纯(氯仿:甲醇=53:47v/v)rf=0.32。旋蒸收集纯品,产率14.70%。
(8)螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn12)的合成
取纯品1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯970mg,于10ml乙腈和水的混合溶液中,搅拌至溶液澄清透明。加入0.22g六水合三氯化钆,室温搅拌2h,析出淡黄色固体,过滤,干燥,柱色谱提纯(氯仿:甲醇=53:47v/v)rf=0.41,产率80.21%。
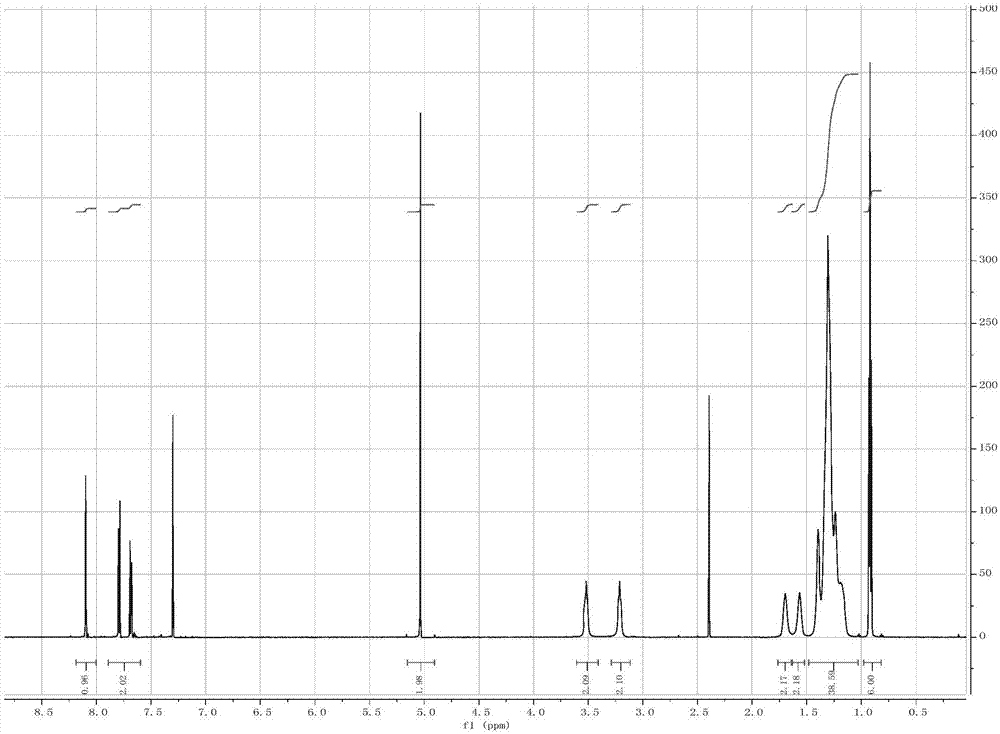
如图7所示,二乙烯三胺五乙酸双酐(dtpaa)的1hnmr图,1h-nmr(600mhz,dmso-d6):δinppm2.56-2.62(t,4h,-n-ch2-ch2-n-);2.70-2.76(t,4h,-n-ch2-ch2-n-);3.28-3.33(s,2h,-n-ch2-cooh);3.68-3.74(s,8h,-cooc-ch2-n-)。
如图8所示,4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺的核磁图,4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺的结构表征如下:1h-nmr(600mhz,cdcl3+dmso-d6):δinppm0.82-0.98(t,6h,-ch2-ch3);1.03-1.48(m,36h,-(ch2)9-ch3);1.52-1.64(m,2h,-conh-ch2-ch2-);1.63-1.76(m,2h,-conh-ch2-ch2-);3.11-3.29(t,2h,-conh-ch2-ch2-);3.41-3.61(t,2h,-conh-ch2-ch2-);4.91-5.15(s,2h,-c6h6-ch2-o-co-);7.60-7.89(dd,2h,-c6h6);8.04-8.15(d,1h,-c6h6-no2)。
如图9所示,1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-烯丙酯的质谱图,目标分子量m=787。m/z822.3848为[m+cl]-峰,m/z1609.6133为[2m+cl]-峰。
如图10所示,1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸4-烯丙酯的核磁图,1h-nmr(600mhz,cdcl3):δinppm0.80-0.93(t,6h,ch2-ch3);1.04-1.38(m,36h,-(ch2)9-ch3);1.42-1.47(s,9h,-(ch3)3);1.46-1.55(t,2h,-conh-ch2-ch2-);1.60-1.73(t,2h,-conh-ch2-ch2-);2.83-3.12(dd,2h,-coo-ch-ch2-coo-),3.13-3.21(t,2h,-conh-ch2-ch2);3.41-3.54(t,2h,-conh-ch2-ch2-);4.65-4.79(q,1h,-nh-ch-coo-);5.18-5.38(dd,2h,-ch=ch2);5.51-5.58(d,1h,-coo-ch-hn-coo);5.59-5.66(s,2h,-c6h6-ch2-);5.78-5.69(m,1h,-ch=ch2);7.60-7.72(dd,2h,-c6h6);8,08-8.17(d,1h,-c6h6-no2)。
1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-丙烯酯的质谱如图11所示,目标分子量m=687。m/z688.5012为[m+h]+峰,m/z=710.4832为[m+na]+峰,m/z=1375.9889为[2m+h]+峰,m/z1397.9740为[2m+na]+峰。
如图12所示,1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-丙烯酯的1hnmr图,1h-nmr(600mhz,cdcl3):δinppm0.73-0.95(t,6h,ch2-ch3);0.97-1.39(m,36h,-(ch2)9-ch3);1.43-1.57(t,2h,-conh-ch2-ch2-);1.58-1.78(t,2h,-conh-ch2-ch2-);2.06-2.23(dd,2h,-coo-ch-ch2-coo-),4.39-4.53(q,1h,-nh-ch-coo-);4.56-4.67(s,2h,-ch2-c2h3-);5.11-5.41(dd,2h,-ch=ch2);5.53-5.69(s,2h,-c6h6-ch2-);5.76-5.95(m,1h,-ch=ch2);7.57-7.77(dd,2h,-c6h6);8,06-8.20(d,1h,-c6h6-no2)。
如图13所示,1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(dtpa-obn12)的质谱如图113所示。目标分子量m=1060,m/z1061.7391为[m+h]+峰。
如图14所示1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯的结构信息如下:1hnmr(600mhz,dmso-d6):δinppm0.73-0.93(t,6h,-ch2-ch3);0.96-1.32(m,36h,-(ch2)9-ch3);1.39-1.50(t,2h,-conh-ch2-ch2-);1.51-1.65(t,2h,-conh-ch2-ch2-);2.76-3.09(t,10h,-n-ch2-ch2-n-,-nh-ch-ch2-coo-);3.21-3.33(t,2h,-conh-ch2-ch2-);3.36-3.48(t,2h,-conh-ch2-ch2-);3.48-3.61(m,8h,-n-ch2-cooh);4.012-4.16(s,2h,-n-ch2-conh);4.47-4.62(s,2h,-ch2-c2h3-);4.80-4.87(q,1h,-nh-ch-coo-);5.11-5.39(dd,2h-ch=ch2);5.45-5.59(s,2h,-c6h6-ch2-);5.81-5.97(m,1h,-ch=ch2);7.64-7.87(dd,2h,-c6h6);8,09-8.11(d,1h,-c6h6-no2);8.55-8.67(s,1h,-ch-nh-co);10.93-13.38(s,4h,n-ch2-cooh)。
如图15所示,由于金属gd具有顺磁性,无法对目标产物进行核磁氢谱表征,通过质谱确定其结构目标分子量m=1217。m/z1216.83为[m-h]-螯合物的一簇峰。
螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸-4-单酰胺五乙酸三胺二乙烯的液相色谱如图16所示,产品含量为98.79%
如图17所示:dtpa-obn12在3300cm-1(v)左右有o-h伸缩振动峰,在1715cm-1(v)左右的c=o伸缩振动峰和1547cm-1(v)仲酰胺n-h的有面内变形振动峰,在1241cm-1(v)处cooh中c-o的伸缩振动峰;而gd-dtpa-obn12在3000cm-1(v)左右有o-h伸缩振动峰减弱,在3400cm-1(v)附近出现宽的结晶水o-h的伸缩振动的峰;1715cm-1(v)左右的c=o伸缩振动峰和1547cm-1(v)仲酰胺n-h的面内变形振动消失;酰胺羰基在1630cm-1(v)左右的伸缩振动峰向低波数迁移;1241cm-1(v)处cooh中c-o的伸缩振动消失,说明配体与金属络合,目标产物正确。
实施例2
(1)二乙烯三胺五乙酸双酐的合成
同实施例1
(2)二乙烯三胺五乙酸单酐的合成
同实施例1
(3)对羧酰氯邻硝基苄溴的合成
取1.00g4-溴甲基-3-硝基苯甲酸于50ml烧瓶中,然后滴加15ml二氯亚砜,回流3h,旋蒸除去未反应二氯亚砜,得淡黄色刺激性气味油状液体4-溴甲基-3-硝基苯酰氯。
(4)4-(溴甲基)-n,n-双十二烷基-3-硝基苯甲酰胺
将0.93g二辛胺和0.6g碳酸钾加入到10ml氯仿中并将所得4-溴甲基-3-硝基苯酰氯溶于20ml干燥氯仿中,逐滴加入到上述溶液中,常温反应12h,水洗除去碳酸钾,氯仿层用无水硫酸钠干燥,除去氯仿,得棕黄色油状液体,柱色谱分离(二氯甲烷:甲醇=10:1v/v),得4-甲基-3-硝基-n,n-二辛基苯酰胺1.53g,产率82.46%。
(5)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸4-烯丙酯的合成
向100ml单口烧瓶中加入干燥好的40ml氯仿,投入1.36g4-(烯丙氧)-2-(叔丁氧基羰基)-4-氧代丁酸,室温下滴加0.82gdbu,室温搅拌30min。升温至90℃,将2.2g4-(溴甲基)-n,n-双辛烷基-3-硝基苯甲酰胺溶于10ml氯仿,向圆底烧瓶中缓慢滴加,滴加结束后回流反应2h。旋干有机溶剂,收集有机相,无水硫酸钠干燥后过滤有机相,旋干液体,得到红棕色的粘稠油状液体,室温下真空干燥12h。柱色谱分离(二氯甲烷:甲醇=10:1v/v)rf=0.65,旋蒸干燥后得到产物1.98g,产率65.20%。
(6)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-烯丙酯的合成
取提纯后的1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)2-(氨基)琥珀酸4-烯丙酯1.0g于50ml圆底单口烧瓶中,加入10ml三氯甲烷于室温搅拌至全部溶解。滴加无水三氟乙酸4g,滴加完成后,搅拌14h,多次旋蒸除去溶剂,30℃真空干燥箱中干燥24h。柱色谱分离(二氯甲烷:甲醇=10:1v/v)rf=0.28,旋蒸干燥后得到产物0.526g,产率95.11%。
(7)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(dtpa-obn8)的合成
将干燥好的0.526g的1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)2-(叔丁氧基羰基)琥珀酸-4-丙烯酯溶于10ml干燥的四氢呋喃,冰水浴下缓慢滴加到上述二乙烯三胺五乙酸单酐液中,保持冰水浴搅拌反应24h。tlc追踪反应,反应完全后,旋干溶剂,沉淀分别用乙醚、乙酸乙酯冲洗,30℃真空干燥。两次柱色谱提纯(氯仿:甲醇=53:47v/v)rf=0.22。旋蒸收集纯品,产率20.70%。
(8)螯合物钆-1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn8)的合成
取纯品1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯210mg,于5ml去甲醇中,搅拌至溶液澄清透明。加入0.12g氧化钆,室温搅拌2h,析出淡黄色絮状固体,过滤,干燥,柱色谱提纯(氯仿:甲醇=53:47v/v)rf=0.28,产率82.67%。
实施例3
(1)乙二胺四乙酸双酐的合成
5.9gedta中加入8ml乙酸酐和12ml无水吡啶,氮气保护,65℃反应24h。反应结束后抽滤,乙酸酐、无水乙醚交替洗涤至白色(淡黄色),85℃真空干燥24h,得固体6.68g(4.76g),产率92.12%。
(2)乙二胺四乙酸单酐的合成
将4.76gedtaa溶于85℃的无水dmf中,加入等摩尔量的去离子水,与80℃加热搅拌2h,后冷却到室温备用。
(3)对羧酰氯邻硝基苄溴的合成
同实施例2
(4)4-(溴甲基)-n,n-双辛烷基-3-硝基苯甲酰胺
同实施例2
(5)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(叔丁氧基羰基)琥珀酸4-烯丙酯的合成同实施例2
(6)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(氨基)琥珀酸-4-烯丙酯的合成
同实施例2
(7)1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(乙二胺四乙酰胺)琥珀酸-4-丙烯酯(edta-obn8)的合成
将干燥好的0.32g的1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)2-(叔丁氧基羰基)琥珀酸-4-丙烯酯溶于10ml干燥的dmso,冰水浴下缓慢滴加到上述乙二胺四乙酸单酐液中,保持冰水浴搅拌反应24h。tlc追踪反应,反应完全后,旋干dmso,分别用乙醚、乙酸乙酯冲洗固体,30℃真空干燥。两次柱色谱提纯(氯仿:甲醇:水=53:45:6v/v)rf=0.28。旋蒸,收集纯品,产率25.14%。
(8)螯合物钆-1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(乙二胺四乙酰胺)琥珀酸-4-丙烯酯(gd-edta-obn8)的合成
取纯品1-(4-(双辛烷基氨基甲酰基)-2-硝基苄基)-2-(乙二胺四乙酰胺)琥珀酸-4-丙烯酯165mg,于5ml甲醇与氯仿的混合溶液中,搅拌至溶液澄清透明。加入0.10g六水合三氯化钆,室温搅拌2h,析出淡黄色絮状固体,过滤,干燥,柱色谱提纯(氯仿:甲醇:水=53:45:6v/v)rf=0.35,产率82.34%。
实施例4
(1)1-(苯甲氧基亚甲基)-二乙烯三胺五乙酸双酐的合成(dtpaa-bsa)
将5gdtpa-bsa,6ml无水乙酸酐,9ml无水吡啶加入到100ml单口烧瓶中,氮气保护下,69℃加热回流,反应24h后,得到深棕色悬浮液,抽滤,滤饼用无水乙酸酐和无水乙醚交替洗至无色,80℃真空干燥24h,得到3.27g白色固体,产率69.13%。
(2)1-(苯甲氧基亚甲基)-二乙烯三胺五乙酸的单酐合成
称取3.27gdtpaa-bsa加入到50ml三口烧瓶中,加入20ml无水dmf,升温至80℃,搅拌使其完全溶解。精确称取等摩尔量的去离子水0.12g于10ml无水dmf中稀释,缓慢滴加到反应溶液中。于90℃下,搅拌反应2.5h,冷却至室温,保存备用。
(3)-(8)同实施例1
实施例5
(1)1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸双酐的合成(dotaa)
将4.04gdota,3ml无水乙酸酐,6ml无水吡啶加入到100ml单口烧瓶中,氮气保护下,69℃加热回流,反应24h后,得到深棕色悬浮液,抽滤,滤饼用无水乙酸酐和无水乙醚交替洗至无色,80℃真空干燥24h,得到2.68g白色固体,产率72.82%。
(2)1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸的单酐合成
称取2.68gdotaa加入到50ml三口烧瓶中,加入20ml无水dmf,升温至80℃,搅拌使其完全溶解。精确称取等摩尔量的去离子水0.13g于10ml无水dmf中稀释,缓慢滴加到反应溶液中。于90℃下,搅拌反应2.5h,冷却至室温,保存备用。
(3)-(8)同实施例1
实施例6
(1)螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn12)空白囊泡的制备方法
称取60.7mggd-dtpa-obn12于样品瓶中,使其溶于5ml甲醇中,配制母液。取母液500μl,氮气旋转吹去溶剂,得到平整脂质薄膜,加入10ml去离子水,然后将混合溶液用超声波信号发生器超声处理10min,得到1mmol/l的囊泡样品溶液。
(2)测试螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn12)的囊泡粒径随光照时间的变化(图1)
配置10ml脂质体溶液,分别取1ml封闭在石英管中,于紫外光下照射0、5、10、20、30、40、60、90、120min。以nanozs90纳米粒度仪,在散射角90o下测量脂质体的粒径分布、光散射强度及分散度。
(3)螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn12)囊泡在光照0min及120min后的tem成像实验(图2)
移取约200μl钆络合物(gd-dtpa-obn12)溶液于封口膜中心,用铜网吸附3min;将铜网取出,用滤纸从铜网侧面吸去残留的液体;晾干后,将铜网浸入磷钨酸(2%)液滴中,负染色3min后,取出铜网,晾干。其形貌通过透射电镜jem-2000ex在电压为120v下进行观测。
(4)螯合物钆-1-(4-(双十二烷基氨基甲酰基)-2-硝基苄基)-2-(二乙烯三胺五乙酰胺)琥珀酸-4-丙烯酯(gd-dtpa-obn12)载药型囊泡的制备方法
取10mmol/l的gd-dtpa-obn12的有机溶剂母液4ml,室温氮气旋转吹去溶剂,得到平整脂质薄膜,真空干燥过夜后,取32.0ml500mg/ldox·hcl的pbs溶液于圆底烧瓶中,然后将混合溶液用超声波发生器超声处理10min。将所得混合溶液,转移到透析袋(mw=3500)中,再将透析袋浸入pbs缓冲液中,并在室温下搅拌,pbs缓冲溶液每0.5h更新一次,透析4h,除去游离阿霉素。
(5)载药型囊泡(dox/gd-dtpa-obn12)的药物包封率的测定
将上述配制好的载药型脂质冷冻干燥除去水,将固体溶于5mldmf中,使用紫外分光光度计(agilen,cary60)测定dmf溶液486nm处的吸光度,与阿霉素的dmf溶液的校准曲线(y=0.018x-0.001,r2=0.999,其中y是486nm处的紫外吸收,x是阿霉素浓度,以mg/l为单位)对比,测定药物包封率(dle),dle根据以下公式计算,gd-dtpa-obn12型脂质的载药含量为67.4%。
(6)载药型囊泡(dox/gd-dtpa-obn12)的粒径随光照时间的变化以及tem形貌观测(图3,图4)
分别取1ml配制好的载药型脂质体溶液封闭在石英管中,于365nm紫外光下照射0、5、10、20、30、40、60、90、120min。以nanozs90纳米粒度仪测量脂质体的粒径分布、光散射强度及分散度。激光器波长为633nm,每次采集15次数据,并对自相关函数进行分析得到结果。取光照0min及120min后的载药脂质溶液约200μl于干净的封口膜中心,使其呈球状液滴,用镊子夹住铜网边缘,使其伸入液体内部,吸附3min;将铜网取出,用滤纸从铜网侧面吸去残留的液体;晾干后,将铜网浸入磷钨酸(2%)液滴中,负染色3min后,取出铜网,晾干后待测。通过透射电镜jem-2000ex在120v电压下观测其形貌。
(7)对比空白gd-dtpa-obn12囊泡与gd-dtpa造影剂的弛豫时间(图5)
将制备好的gd-dtpa-obn12脂质溶于pbs缓冲溶液(0.01m;ph7.4)中,配制成浓度稍高的母液,用icp-aes测定钆离子浓度,再梯度稀释到一定浓度,置于5ml离心管中,通过0.54t微型核磁共振成像仪,在37℃下,利用反转恢复法,测定不同钆离子浓度的纵向弛豫时间(t1)。测得弛豫时间与钆离子浓度拟合直线斜率即为纵向弛豫效率(r1)。
(8)对比空白gd-dtpa-obn12囊泡与gd-dtpa造影剂的体外造影成像效果(图6)
将制备好的gd-dtpa-obn和gd-dtpa溶于pbs缓冲溶液中,配制成浓度稍高的母液,用icp-aes测定钆离子浓度,再分别稀释到1.25,1.05,0.85,0.65,0.45,0.25,0.15,0.10mmol/l,置于离心管中,由0.54t微型核磁共振成像仪37℃下进行体外核磁共振成像。
- 还没有人留言评论。精彩留言会获得点赞!