一种N-苯乙酰基-L-脯氨酸的制备方法与流程
本发明属于有机合成技术领域,涉及一种药物中间体的制备方法,特别是涉及一种n-苯乙酰基-l-脯氨酸的制备方法。
背景技术:
n-苯乙酰基-l-脯氨酸是一种酰胺类化合物,酰胺类化合物在药物化学、高分子化学以及生物化学领域应用广阔是有机合成中重要的化合物之一。研究表明绝大部分的药物属于酰胺类化合物,因此酰胺类化合物的制备在药物合成方面有很好的前景,而且n-苯乙酰基-l-脯氨酸是合成营养强化剂n-[1-(苯基乙酰基)-l-脯氨酰]甘氨酸乙酯的重要中间体。
1996年gudasheva等在其文章中首先提到利用氢氧化钠水溶液将l-脯氨酸变为l-脯氨酸钠盐,再以氢氧化钠作为缚酸剂在水中与其钠盐进行反应制备出n-苯乙酰基-l-脯氨酸钠盐,最后经酸化得产品,收率86%,(详见gudasheva;voronina;ostrovskaya;rozantsev;vasilevich;trofimov;kravchenko;skoldinov;seredenin;europeanjournalofmedicinalchemistry;vol.31;nb.2;(1996);p.151-157)。该方法不仅操作复杂,而且在水相中反应引起部分苯乙酰氯的水解,造成副反应多,产品纯度低,杂质多。
2005年boto,alicia等人在其文章中提到在四氢呋喃中以饱和碳酸氢钠水溶液作为缚酸剂,在水溶液和四氢呋喃的混合溶剂中室温下搅拌反应10小时制备n-苯乙酰基-l-脯氨酸,收率76%,(详见synthesisofalkaloidanaloguesfromα-aminoacidsbyone-potradicaldecarboxylation/alkylation.byboto,aliciaetalfromeuropeanjournaloforganicchemistry,(16),3461-3468;2005.)。该方法操作简单,但收率低,产品的精制提纯较困难。
2014年严建祥在201410028081.6的n-苯乙酰-l-脯氨酸和n-[1-(苯基乙酰基)-l-脯氨酰]甘氨酸乙酯的制备方法同样公开了在水体系中进行反应,先将l-脯氨酸与氢氧化钠水溶液反应使其变为l-脯氨酸钠盐,l-脯氨酸钠盐继续与苯乙酰氯进行酰胺化反应获得n-苯乙酰基-l-脯氨酸钠盐,最后经酸化得产品,反应时间短反应温度低,副反应多,反应不彻底,而且副产物污染环境。
2016年zielinska,katarzyna等在其文章中提到了α-氨基酸的苯乙酰化作用,与严建祥等人制备方法相同,均是在水中先将氨基酸变为氨基酸的钠盐,再以氢氧化钠水溶液作为缚酸剂在水中进行反应制备出n-苯乙酰基-l-脯氨酸钠盐,在进行酸化得产品(详见zielińska,katarzyna;mazurkiewicz,roman;szymańska,katarzyna;jarzębski,andrzej;magiera,sylwia;erfurt,karol;journalofmolecularcatalysisb:enzymatic;vol.132;(2016);p.31-40)。制备操作复杂,副产物多,并未报道其收率。
技术实现要素:
为了解决现有技术中产品纯度低,提纯困难及收率低的问题,本发明公开了一种n-苯乙酰基-l-脯氨酸的制备方法,在无水有机溶剂中催化剂作用下l-脯氨酸与苯乙酰氯直接进行反应,避免了苯乙酰氯的水解,反应更加高效彻底,产品收率高,合成工艺简单,操作简单,易于提纯,产品纯度较高。
本发明通过以下实验方案来实现:
本发明公开了一种n-苯乙酰基-l-脯氨酸的制备方法,在有机溶剂体系中,以l-脯氨酸为原料,苯乙酰氯为酰化剂,在相转移催化剂的作用下通过酰胺化反应制备得到n-苯乙酰基-l-脯氨酸。
本发明合成路线如下:
进一步地,上述n-苯乙酰基-l-脯氨酸的制备方法,具体是通过如下步骤实现的:
(1)低温下,向溶解有l-脯氨酸的有机溶剂中滴加苯乙酰氯及缚酸剂,滴加20-40min后,加入相转移催化剂,搅拌反应1-9h后升温至20-80℃继续搅拌反应2-8h,得到反应液;其中缚酸剂分批次缓慢加入效果更佳;优选地,所述低温条件为-5-5℃。
(2)将反应液趁热过滤,滤液水洗,有机相干燥,旋蒸去除溶剂得到白色固体,所得白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸,其熔点为150.3-151.7℃,与文献报道值150-152℃相吻合。
作为一种优选实施方式,反应物中l-脯氨酸、苯乙酰氯、缚酸剂物质的量之比为(1-2):(1-3):(3-5),所述相转移催化剂的用量占反应物的摩尔分数为(0.5-10)mol%。
作为一种优选实施方式,所述缚酸剂选自na2co3、k2co3、n,n-二异丙基乙胺(diea)、吡啶、三乙胺中的一种。
作为一种优选实施方式,所述有机溶剂选自四氢呋喃、氯仿、丙酮、二甲基亚砜、苯、n,n-二甲基甲酰胺中的一种。
作为一种优选实施方式,所述有机溶剂提前利用无水硫酸钠或者无水硫酸镁干燥。
作为一种优选实施方式,所述相转移催化剂选自聚乙二醇-400(peg-400)、环糊精、四丁基溴化铵(tbab)、十二烷基三甲基氯化铵(dtac)、三乙基苄基氯化胺(teba)中的一种。
本发明的优点:
(1)本发明以l-脯氨酸为原料,苯乙酰氯为酰化剂,原料易得,成本低;
(2)本发明在干燥过的无水有机溶剂中进行反应,l-脯氨酸与苯乙酰氯在催化剂作用下直接进行酰胺化反应制备n-苯乙酰基-l-脯氨酸,与前人报道的其在水相中反应机理不同。采用干燥过的无水溶剂有效的避免了苯乙酰氯的水解。并且未经无水硫酸镁干燥的有机溶剂自身带有一部分水份,同样会造成少部分苯乙酰氯的水解,在实际生产应用中避免了其水解对原料的浪费,降低了生产成本;本发明采用无水条件成功的避免了副反应的发生,使得反应更加彻底,有效的提高了其收率,且本发明l-脯氨酸与苯乙酰氯直接反应得到产品,无需酸化,产品分离简单,纯度高;
(3)本发明通过使用相转移催化剂来加快反应速率,反应收率高达98.5%。tbab、dtac以及teba均属于季铵盐类r4n+x-相转移催化剂,在固液相反应过程中,季铵盐正离子q+与试剂l-脯氨酸负离子结合成离子对,并利用正离子q+自身的亲酯性将l-脯氨酸负离子带入有机相中,在有机相中试剂负离子不能溶剂化,从而大大提高了试剂负离子的反应活性,从而提高了反应速率和收率。peg-400属于聚醚类相转移催化剂,在反应过程中peg-400形成6-8个氧原子处于同一平面的假环状结构,分子链的其余部分在平面的一侧形成一个络合正离子,络合正离子与试剂负离子结合成离子对进入有机相,使试剂负离子裸露在有机相中,从而以较高的反应活性参与反应;
(4)相比前人报道的先制得n-苯乙酰-l-脯氨酸钠盐再经稀盐酸酸化获得产品,本发明采用固液相直接法进行反应,合成工艺简单,操作容易,其次原料直接进行酰胺化反应有效的避免了副反应的发生,最后旋蒸除去溶剂再经重结晶既得产品n-苯乙酰基-l-脯氨酸,避免了钠盐酸化过程中产生的副产物,提纯简单,产品纯度较高。
附图说明
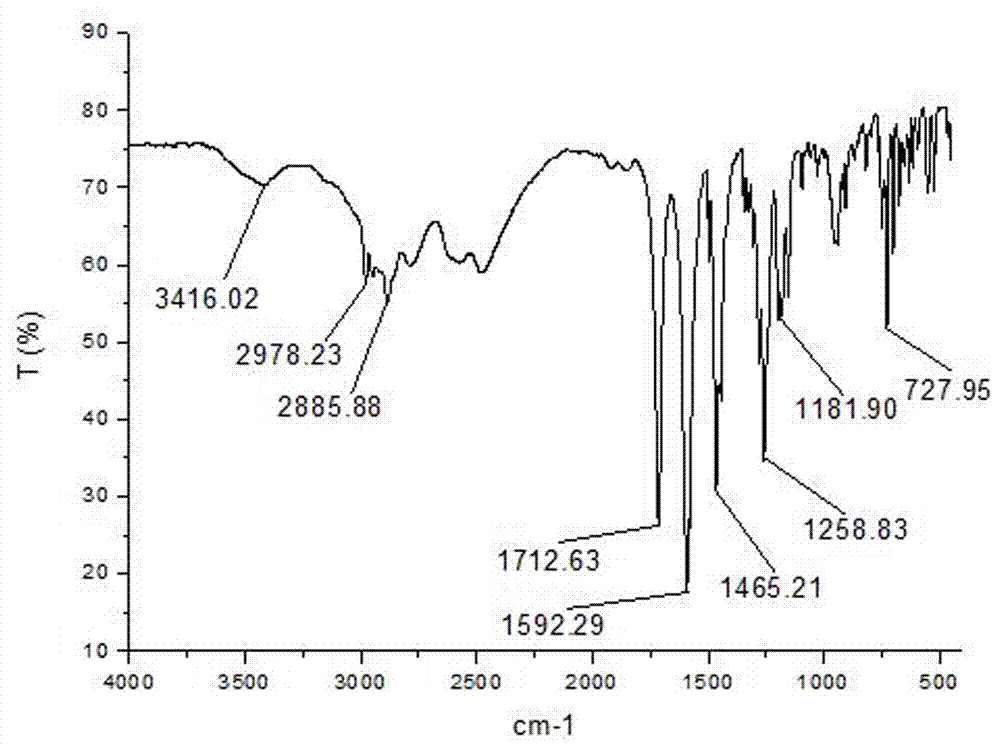
图1是实施例1制备得到的n-苯乙酰基-l-脯氨酸的红外光谱图ir(kbr,vcm-1)。
图2是实施例1制备得到的n-苯乙酰基-l-脯氨酸的核磁共振氢谱1hnmr(cdcl3)δ。
具体实施方式
实施例1
分别称取3g(0.026mol)l-脯氨酸,12.058g(0.078mol)苯乙酰氯,7.18g(0.052mol)k2co3备用,将40ml干燥过的丙酮和l-脯氨酸加入装有干燥管、温度计、机械搅拌、冷凝管的100ml三口瓶中,冰水浴-2-0℃搅拌下向三口瓶中用针管缓慢滴加苯乙酰氯,期间分批加入k2co3缚酸剂,继续低温0℃下加入5mol%peg-400,保温搅拌反应1h,换油锅20℃加热搅拌6h,反应液趁热过滤,滤液水洗,有机相干燥,减压蒸馏得白色固体,白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸,其熔点为150.5-151.4℃,收率为97.8%。
将实施例1中所得白色固体进行红外色谱和核磁共振检测。
由图1的红外谱图可以看出:3416.02cm-1处为羧基中o-h键吸收峰;2978.23cm-1,2885.88cm-1处为脯氨酸环上亚甲基-ch2-吸收峰;1712.63cm-1处为羧基c=o键吸收峰;1592.29cm-1处为叔酰胺中c=o键吸收峰;在1465.21cm-1处为-ch2-吸收峰;1258.83cm-1处为羧基中c-o键吸收峰;在1181.90cm-1处为叔酰胺中c-n键吸收峰;727.95cm-1处为苯环单取代特征峰。综上所述,说明白色固体为n-苯乙酰基-l-脯氨酸。
由图2的核磁共振氢谱图可以看出2.10为脯氨酸环相连两个亚甲基上的4个h;3.48,3.59为脯氨酸环上连接叔酰胺的2个h;3.75为苯乙酰基上亚甲基的2个h;4.63为脯氨酸上连接羧基处的1个h;7.25-7.37为苯乙酰基上苯环5个h。进一步说明了所得产品为n-苯乙酰基-l-脯氨酸。
实施例2
分别称取4.49g(0.039mol)l-脯氨酸,4.02g(0.026mol)苯乙酰氯,15.12g(0.117mol)n,n-二异丙基乙胺(diea)备用,将40ml干燥过的苯和l-脯氨酸加入装有干燥管、温度计、机械搅拌、冷凝管的100ml三口瓶中,冰水浴0-5℃搅拌下向三口瓶中用针管缓慢滴加苯乙酰氯,同时分批加入diea缚酸剂,继续低温0℃下加入0.5mol%四丁基溴化铵(tbab),保温搅拌反应7h,换油锅升温80℃回流2h,反应液趁热过滤,滤液水洗,有机相干燥,减压蒸馏得白色固体,所得白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸,其熔点为150.4-151.6℃,收率98.5%。
实施例3
分别称取3g(0.026mol)l-脯氨酸,8.04g(0.052mol)苯乙酰氯,10.28g(0.13mol)吡啶备用,将40ml干燥过的氯仿和l-脯氨酸加入装有干燥管、温度计、机械搅拌、冷凝管的100ml三口瓶中,冰水浴-5-3℃搅拌下向三口瓶中用针管缓慢滴加苯乙酰氯,同时分批加入吡啶缚酸剂,继续低温0℃下加入2.5mol%环糊精,保温搅拌反应5h,换油锅升温60℃回流4h,反应液趁热过滤,滤液水洗,有机相干燥,减压蒸馏得白色固体,所得白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸。熔点为150.4-151.7℃,收率98.4%。
实施例4
分别称取6g(0.052mol)l-脯氨酸,12.06g(0.078mol)苯乙酰氯,9.21g(0.091mol)三乙胺备用,将40ml干燥过的四氢呋喃和l-脯氨酸加入装有干燥管、温度计、机械搅拌、冷凝管的100ml三口瓶中,冰水浴-2-4℃搅拌下向三口瓶中用针管缓慢滴加苯乙酰氯,期间分批加入三乙胺缚酸剂,继续低温0℃下加入10mol%氯化三乙基苄基胺(teba),保温搅拌反应3h。换油锅升温40℃搅拌反应8h,反应液趁热过滤,滤液水洗,有机相干燥,减压蒸馏得白色固体,所得白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸。熔点为150.3-151.5℃,收率97.4%。
实施例5
分别称取6g(0.052mol)l-脯氨酸,4.028g(0.026mol)苯乙酰氯,10.77g(0.078mol)na2co3备用,将40ml干燥过的n,n-二甲基甲酰胺和l-脯氨酸加入装有干燥管、温度计、机械搅拌、冷凝管的100ml三口瓶中,冰水浴0-5℃搅拌下向三口瓶中用针管缓慢滴加苯乙酰氯,期间分批加入k2co3缚酸剂,加料完成,保温搅拌反应9h,换油锅升温60℃搅拌反应2h,反应液趁热过滤,滤液水洗,有机相干燥,减压蒸馏得白色固体,所得白色固体重结晶即得产品n-苯乙酰基-l-脯氨酸。熔点为150.5-151.2℃,收率90.6%。
- 还没有人留言评论。精彩留言会获得点赞!