人工合成的polyA、哺乳动物细胞重组表达载体、哺乳动物宿主细胞、表达方法及应用与流程
本发明涉及一种人工合成的polya、哺乳动物细胞重组表达载体、哺乳动物宿主细胞、表达方法及应用,属于生物技术领域。
背景技术:
重组蛋白在细菌、真核微生物、昆虫细胞等宿主中的表达虽然能获得较高的产量,但是这些表达系统缺少类似于哺乳动物细胞的翻译后蛋白质修饰机制的功能,例如准确的n型糖基化和o型糖基化等,这会导致表达的蛋白缺少生物活性或因折叠错误而产生包涵体。与其他系统相比,哺乳动物表达系统的优势在于能够指导蛋白质的正确折叠,提供复杂的糖基化等多种翻译后加工功能,正因为这些优势的存在使得表达产物在分子结构、理化性质和生物学功能方面非常接近于天然的高等生物蛋白质分子。
外源蛋白质在哺乳动物细胞中的表达受很多因素的影响,如表达载体的调控元件、转录和翻译控制因子、转基因拷贝数、mrna的稳定性、外源基因在染色体上的整合位点、重组蛋白对宿主细胞的潜在毒性和宿主细胞的基因组偏好性等。多聚腺苷酸,即polya,普遍存在形式是mrna的polya尾。几乎所有真核mrna的加工过程中,mrna的3'末端都有polya尾,参与许多mrna代谢过程。另外mrna的3'端的polya尾能够调控mrna的稳定性,提高mrna的翻译效率,在成熟mrna从细胞核向细胞质转运过程中起着重要作用。此外,polya能保护mrna,免受核酸外切酶攻击,并且影响转录终止、蛋白质翻译。
目前,对提高哺乳动物细胞的表达载体的优化主要集中在启动子、增强子、核基质附着区等,而polya元件对哺乳动物细胞转基因表达的影响并未见报道。
技术实现要素:
本发明的目的是提供一种人工合成的polya,该人工合成的polya可以增强mrna的稳定性,进而提高哺乳动物宿主细胞中外源蛋白的表达。
本发明还提供一种哺乳动物细胞重组表达载体,该哺乳动物细胞重组表达载体含上述人工合成的polya,可以提高外源蛋白质在哺乳动物细胞中的表达量。
另外,本发明提供一种哺乳动物宿主细胞,该哺乳动物宿主细胞含上述哺乳动物表达载体,可以改善哺乳动物细胞外源蛋白表达量低的问题。
此外,本发明还提供一种哺乳动物细胞表达外源蛋白质的表达方法,通过该方法获得的目的蛋白表达水平高,生产成本低。
本发明还提供上述人工合成的polya、哺乳动物细胞重组表达载体、或者哺乳动物宿主细胞在提高哺乳动物细胞重组蛋白表达水平中的应用。该应用可以提高mrna的翻译效率,增强动物细胞中mrna的稳定性,提高外源蛋白的表达。
为了实现上述目的,本发明所采用的技术方案是:
一种人工合成的polya,所述人工合成的polya的核苷酸序列如seqidno.1所示。该核苷酸序列可以成倍提高外源蛋白在哺乳动物细胞的表达量。
一种哺乳动物细胞重组表达载体,包含哺乳动物来源的蛋白质序列,所述哺乳动物细胞重组表达载体还含有如seqidno.1所示的人工合成的polya的核苷酸序列。该哺乳动物细胞重组表达载体可以使egfp基因表达量提高1倍多,使阿达木抗体表达量提高1~2倍,进而降低了哺乳动物细胞生产重组蛋白的成本。
所述哺乳动物细胞重组表达载体中的启动子为cmv、sv40、ef-1α、cag中的任意一种。上述启动子为哺乳动物细胞重组表达载体常用的启动子,适用于哺乳动物细胞重组表达载体中。
所述启动子为cmv启动子。cmv启动子是哺乳动物细胞重组表达载体中最常用的且表达量比较高的启动子。
一种哺乳动物宿主细胞,所述哺乳动物宿主细胞由上述哺乳动物细胞重组表达载体转染得到。该哺乳动物宿主细胞中含上述哺乳动物细胞重组表达载体,
所述哺乳动物宿主细胞为cho、hek293、bhk或sp2/o。所述哺乳动物宿主细胞为重组蛋白表达系统的常用细胞系,其培养方式简单,易于大规模培养。
一种哺乳动物细胞表达外源蛋白质的表达方法,包括以下步骤:
(1)构建包含哺乳动物外源蛋白质序列以及如seqidno.1所示的人工合成的polya的核苷酸序列的重组表达载体;
(2)将重组表达载体转入哺乳动物宿主细胞中;
(3)培养步骤(2)中的哺乳动物宿主细胞,表达目的蛋白。
通过该方法操作简便,成本低,通过该方法获得的目的蛋白表达量成倍提升,还可以增强转录产物的稳定性。
上述人工合成的polya、哺乳动物细胞重组表达载体、或者哺乳动物宿主细胞在提高哺乳动物细胞重组蛋白表达水平中的应用。该应用可以使哺乳动物来源蛋白表达量提高一倍以上。
附图说明
图1为本发明试验例1中重组表达载体与对照载体瞬时转染率对比图;
图2为本发明试验例1中的重组表达载体与对照载体egfp平均表达量对比图;
图3为本发明试验例2中的重组表达载体与对照载体阿达木抗体平均表达量对比图。
具体实施方式
下面结合附图对本发明的实施方式作进一步说明,但不构成对本发明的任何限制。除所列举的实施例外,还可以有其他的实施方式,但是凡采用等同替换或等效变换获得的技术方案,均落在本发明要求的保护范围内。
实施例及试验例中所用的质粒小提试剂盒购自康为世纪生物科技有限公司(货号:cw0500s);无缝克隆试剂盒购自碧云天生物公司(货号:d7010s),工具酶、培养基等均为市售商品;pires-neo、pegfp-c1质粒购自clontech生物公司;下述实施例中人工合成的polya由通用生物基因(安徽)有限公司完成合成。
人工合成的polya的实施例
本实施例的人工合成的polya,命名为polya-1,核苷酸序列如下所示:
cagacatgataagatacattgatgagtttggacaaaccacaactagaatgcagtgaaaaaaatgctttatttgtgaaatttgtgatgctattgctttatttgtaaccattataagctgcaataaacaagttaacaacaacaattgcattcattttatgtttcaggttcagggggaggtgtgggaggttttttaaagcaagtaaaacctctacaaatgtggta。(seqidno.1)
哺乳动物细胞重组表达载体的实施例
本实施例为含有人工合成的polya-1的重组表达载体的构建,包括以下步骤:
(1)egfp基因扩增
参照pegfp-c1载体的enhancedgreenfluorescentprotein(egfp)基因序列(genbank:u55763.1,第613~1410位碱基)设计上游引物p-f和下游引物p-r,引物的5′端分别引入ecori、bamhi酶切位点,引物序列如下所示(下划线处为酶切位点):
p-f:5′-ccggaattcatggtgagcaagggcgaggag-3′;
p-r:5′-ctaggatccttactagatccggtggatcc-3′。
以pegfp-c1质粒(购自美国clontech公司)为模板,使用引物p-f、p-r扩增egfp基因。pcr反应体系如下表1所示。
表1pcr反应体系
反应程序:95℃预变性3min;94℃变性40s,58℃退火30s,72℃延伸40s,共30个循环;72℃延伸3min;4℃保存。
琼脂糖凝胶电泳回收pcr扩增产物,纯化后送生物公司测序验证。结果表明,扩增出的dna片段与genbank公开的egfp序列完全一致。
(2)构建含人工合成的polya-1的表达载体
采用无缝克隆试剂盒将pires-neo载体的polya核苷酸序列替换polya-1(seqidno.1)构成新的表达载体;用smai/paci酶对新的表达载体进行双酶切验证,酶切验证酶切体系为:cutsmartbuffer2μl,先加入smai酶0.5μl(浓度为10u/μl),0.81μg/μl的质粒dna1.23μl,补足水至20μl。充分混匀后,25℃孵育3h;再加入paci酶0.5μl(浓度为10u/μl),充分混匀后,37℃孵育3h。核酸电泳验证,酶切条带大小正确的表达载体再进行测序验证,构建正确的表达载体,命名为pires-1。
(3)构建含egfp序列和表达载体pires-1的重组表达载体
用ecori、bamhi双酶切egfp的pcr扩增产物(经测序验证正确的序列),同时用ecori、bamhi双酶切上述表达载体pires-1的dna,琼脂糖凝胶电泳鉴定酶切结果,凝胶回收酶切后的egfp序列片段和表达载体pires-1的dna。
egfp序列的酶切体系为:10×mbuffer2μl,10u/μlecori、bamhi酶各0.5μl,1.289μg/μlegfp扩增产物0.78μl,补足水至20μl。充分混匀后,37℃孵育6h。
表达载体pires-1的酶切体系为:10×kbuffer2μl,10u/μlecori、bamhi酶各0.5μl,0.81μg/μlpires-1的dna1.23μl,补足水至20μl。充分混匀后,37℃孵育3h。
用t4连接酶连接酶切后的egfp序列片段和pires-1。
表达载体pires-1的连接体系为:2×quickligationbuffer10μl,pires-1的线性dna200ng,酶切后的egfp序列片段87.2ng,350u/μl的t4连接酶1μl,补足水至20μl,16℃连接过夜。
将连接产物加入大肠杆菌(e.coli)jm109感受态细菌悬液中转化,取100μl转化菌液接种在含有氨苄青霉素的lb固体培养板上,37℃培养过夜,挑取单菌落摇菌继代培养。提取细菌质粒,用ecori和bamhi双酶切细菌质粒,酶切体系为:10×kbuffer2μl,10u/μl的ecori、bamhi酶各0.5μl,0.81μg/μl连接载体的dna1.23μl,补足水至20μl。充分混匀后,37℃孵育3h。核酸电泳验证,酶切正确的条带大小为720bp,选取酶切鉴定正确的质粒,进行测序验证,最后获得正确的重组表达载体,命名为pires-e1。
哺乳动物宿主细胞的实施例
本实施例哺乳动物宿主细胞的实施例,包括以下步骤:
cho细胞于37℃、5%co2条件下,在含10%灭活胎牛血清的dmem培养基中培养。在6孔板内接种cho细胞(3×106/孔)。铺板培养24h后细胞达到约90%融合度,将哺乳动物细胞重组表达载体的实施例中的重组表达载体pires-e1转染进入cho细胞中,转染24h后,用浓度为800μg/ml的g418筛选2周出现阳性克隆时,将浓度为800μg/ml的g418更换为400μg/ml浓度的g418稳定培养1个月,即得哺乳动物宿主细胞cho。
哺乳动物来源蛋白质的表达方法的实施例
本实施例哺乳动物来源蛋白质的表达方法,包括以下步骤:
(1)构建包含egfp基因序列以及人工合成的polya-1的核苷酸序列的重组表达载体,构建方法和上述哺乳动物细胞重组表达载体的实施例的构建方法相同;
(2)将重组表达载体转染进入哺乳动物宿主细胞cho中,转染方法和上述哺乳动物宿主细胞的实施例中的转染方法相同;
(3)培养cho细胞,表达目的蛋白。
人工合成的polya、哺乳动物细胞重组表达载体、哺乳动物宿主细胞在提高哺乳动物细胞重组蛋白表达水平中的应用的实施例
本实施例重组表达载体pires-e1的构建方法与哺乳动物细胞重组表达载体的实施例中重组表达载体pires-e1的构建方法完全相同;哺乳动物宿主细胞cho的转染方法与哺乳动物宿主细胞的实施例中哺乳动物宿主细胞cho的转染方法完全相同,将转染后的宿主细胞cho进行流式细胞检测,报告基因egfp的表达量。
人工合成的polya的对比例1
本对比例的人工合成的polya,命名为polya-1′,核苷酸序列如下所示:
polya-1′:
ctgtgccttctagttgccagccatctgttgtttgcccctcccccgtgccttccttgaccctggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtctgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagggggaggattgggaagacaatagcaggcatgctggggatgcggtgggctctatgg。(seqidno.2)
人工合成的polya的对比例2
本对比例的人工合成的polya,命名为polya-2′,核苷酸序列如下所示:
aataaaagatctttattttcattagatctgtgtgttggttttttgtgtg。(seqidno.3)
人工合成的polya的对比例3
本对比例的人工合成的polya,命名为polya-3′,核苷酸序列如下所示:
aataaaaagacagaataaa。(seqidno.4)
哺乳动物细胞重组表达载体的对比例1
本对比例采用无缝克隆试剂盒将pires-neo载体的polya核苷酸序列替换为polya-1′(seqidno.2)构成新的表达载体;用smai/paci酶对新的表达载体进行双酶切验证,取酶切验证正确的载体再进行测序验证,构建正确的表达载体,命名为pires-1′。
本对比例的哺乳动物细胞重组表达载体的构建方法同哺乳动物细胞重组表达载体的实施例,仅将pires-1替换为pires-1′,构建正确的重组表达载体命名为pires-e1′。
哺乳动物细胞重组表达载体的对比例2
本对比例采用无缝克隆试剂盒将pires-neo载体的polya核苷酸序列替换为polya-2′(seqidno.3)构成新的表达载体;用smai/paci酶对新的表达载体进行双酶切验证,取酶切验证正确的载体再进行测序验证,构建正确的表达载体,命名为pires-2′。
本对比例的哺乳动物细胞重组表达载体的构建方法同哺乳动物细胞重组表达载体的实施例,仅将pires-1替换为pires-2′,构建正确的重组表达载体命名为pires-e2′。
哺乳动物细胞表达载体的对比例3
本对比例采用无缝克隆试剂盒将pires-neo载体的polya核苷酸序列替换为polya-3′(seqidno.4)构成新的表达载体;用smai/paci酶对新的表达载体进行双酶切验证,取酶切验证正确的载体再进行测序验证,构建正确的表达载体,命名为pires-3′。
本对比例的哺乳动物细胞重组表达载体的构建方法同哺乳动物细胞重组表达载体的实施例,仅将pires-1替换为pires-3′,构建正确的重组表达载体命名为pires-e3′。
哺乳动物宿主细胞的对比例1
本对比例的哺乳动物宿主细胞与哺乳动物宿主细胞的实施例中的哺乳动物宿主细胞的区别仅在于,将哺乳动物宿主细胞的实施例中的重组表达载体pires-e1替换为哺乳动物细胞重组表达载体的对比例1中的重组表达载体pires-e1′。
哺乳动物宿主细胞的对比例2
本对比例的哺乳动物宿主细胞与哺乳动物宿主细胞的实施例中的哺乳动物宿主细胞的区别仅在于,将哺乳动物宿主细胞的实施例中的重组表达载体pires-e1替换为哺乳动物细胞重组表达载体的对比例2中的重组表达载体pires-e2′。
哺乳动物宿主细胞的对比例3
本对比例的哺乳动物宿主细胞与哺乳动物宿主细胞的实施例中的哺乳动物宿主细胞的区别仅在于,将哺乳动物宿主细胞的实施例中的重组表达载体pires-e1替换为哺乳动物细胞重组表达载体的对比例3中的重组表达载体pires-e3′。
试验例1
本试验例将哺乳动物细胞重组表达载体的实施例中的重组表达载体pires-e1、哺乳动物细胞重组表达载体的对比例1中的重组表达载体pires-e1′、哺乳动物细胞重组表达载体的对比例2中的重组表达载体pires-e2′、哺乳动物细胞重组表达载体的对比例3中的重组表达载体pires-e3′分别转染进入cho细胞,具体步骤如下:
(1)细胞转染
cho细胞于37℃、5%的co2条件下,在含10%灭活胎牛血清的dmem培养基中培养。在6孔板内接种cho细胞(3×106/孔)。铺板培养24h后细胞达到约90%融合度。试验共分为6组:①正常cho细胞组;②对照组-转染对照载体pires;③转染重组表达载体pires-e1;④转染重组表达载体pires-e1′;⑤转染重组表达载体pires-e2′;⑥转染重组表达载体pires-e3′。以lip3000
(2)稳定转染细胞系的筛选
转染24h后,再将各试验组载体分别转染培养于24孔板(500μl/well)的cho细胞中(cho的细胞个数约为2×106个),用浓度为800μg/ml的g418筛选2周出现阳性克隆时,将浓度为800μg/ml的g418更换为400μg/ml浓度的g418稳定培养1个月,然后收集各试验组细胞进行流式细胞检测。
(3)细胞转染的瞬时表达分析
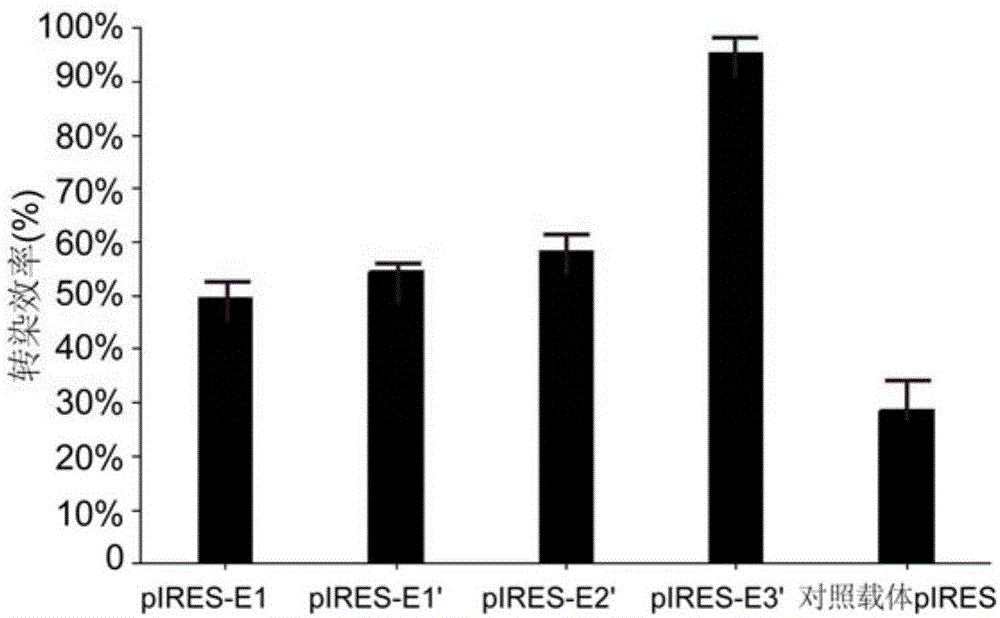
转染48h后分析结果,其中,正常cho细胞作为空白对照筛选阳性克隆,筛选出阳性克隆后正常cho细胞已被杀死。转染结果如图1所示,对照载体pires的转染效率为28.7%;重组表达载体pires-e1的转染效率为95.3%;重组表达载体pires-e1′的转染效率为49.5%;重组表达载体pires-e2′的转染效率为54.6%;重组表达载体pires-e3′的转染效率为58.4%。
(4)egfp表达检测
流式细胞检测报告基因egfp的表达量,结果如图2所示,在相同的条件下,本发明的重组表达载体pires-e1与对照载体pires相比,egfp的平均表达量提高了1.3倍。
试验例2
本试验例中阿达木抗体重链或轻链的合成由通用生物基因(安徽)有限公司完成合成。
1、含阿达木抗体的重组表达载体的构建
用ecori/bamhi双酶切阿达木重链或轻链序列的pcr扩增产物(经测序验证正确的序列),同时用ecori/bamhi双酶切egfp表达量较好的重组表达载体pires-e1对应的表达载体pires-1和pires-e2′对应的表达载体pires-2′的dna。琼脂糖凝胶电泳鉴定酶切结果,凝胶回收酶切后的阿达木重链或轻链序列片段和pires-1、pires-2′的线形质粒dna。
阿达木重链或轻链序列的双酶切体系为:阿达木重链或轻链序列片段10μl(1μg/μl),10×nebuffer2.13μl,ecori/bamhi酶(10u/μl)各1.0μl,补足水至30μl;酶切条件为:37℃,酶切3min。
pires-1的双酶切体系为:pires-15μl(1μg/μl),10×nebuffer2.12μl,ecori/bamhi(10u/μl)各0.5μl,补足水至20μl;酶切条件为:37℃,酶切3min。
pires-2′的双酶切体系为:pires-2′5μl(1μg/μl),10×nebuffer2.12μl,ecori/bamhi(10u/μl)各0.5μl,补足水至20μl;酶切条件为:37℃,酶切3min。
取酶切后的阿达木重链或轻链序列片段和pires-1、pires-2′线形质粒dna(摩尔比5:1),使用neb公司tm的连接试剂盒,25℃连接5min。将连接产物加入到大肠杆菌(e.coli)jm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组表达载体并进行双酶切(ecori/bamhi)验证,取酶切验证正确的载体进行测序验证,最后获得正确的重组表达载体,含表达载体pires-1、pires-2′的重组表达载体分别命名为pires-p1、pires-p2′。
2、细胞转染及稳定转染细胞系的筛选
(1)细胞转染
cho细胞于37℃、5%co2条件下,在含10%灭活胎牛血清的dmem培养基中培养。在6孔板内接种cho细胞(3×106/孔)。铺板培养24h后细胞达到约90%融合度。试验共分为4组:①正常cho细胞组;②对照组-转染对照载体pires;③转染重组载体pires-p1;④转染重组载体pires-p2′。以lip3000
(2)稳定转染细胞系的筛选
载体共转染培养于24孔板(500μl/well)的cho细胞中(cho的细胞个数约2×106个),用浓度为800μg/ml的g418筛选2周,当阳性克隆出现时,换浓度为400μg/ml的g418稳定培养2周后,每毫升接种细胞量为50~60万个,摇床转速设置为120rpm,悬浮培养第7天收集培养液上清。
3、阿达木抗体表达量检测
用elisa试剂盒检测阿达木抗体表达量。结果如图3所示,与对照组相比,含重组表达载体pires-p1的阿达木抗体提高了1.4倍,即本发明的人工合成的polya的可以提高外源蛋白在动物细胞中的表达量。
<110>新乡医学院、河南普诺易生物制品研究院有限公司
<120>人工合成的polya、哺乳动物细胞重组表达载体、哺乳动物宿主细胞、表达方法及应用
<160>6
<170>siposequencelisting1.0
<211>222
<212>dna
<213>人工序列
<400>1
cagacatgataagatacattgatgagtttggacaaaccacaactagaatgcagtgaaaaa60
aatgctttatttgtgaaatttgtgatgctattgctttatttgtaaccattataagctgca120
ataaacaagttaacaacaacaattgcattcattttatgtttcaggttcagggggaggtgt180
gggaggttttttaaagcaagtaaaacctctacaaatgtggta222
<211>225
<212>dna
<213>人工序列
<400>2
ctgtgccttctagttgccagccatctgttgtttgcccctcccccgtgccttccttgaccc60
tggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtc120
tgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagggggaggatt180
gggaagacaatagcaggcatgctggggatgcggtgggctctatgg225
<211>49
<212>dna
<213>人工序列
<400>3
aataaaagatctttattttcattagatctgtgtgttggttttttgtgtg49
<211>19
<212>dna
<213>人工序列
<400>4
aataaaaagacagaataaa19
<211>30
<212>dna
<213>人工序列
<221>上游引物p-f
<400>5
ccggaattcatggtgagcaagggcgaggag30
<211>29
<212>dna
<213>人工序列
<221>上游引物p-r
<400>6
ctaggatccttactagatccggtggatcc29
- 还没有人留言评论。精彩留言会获得点赞!