普罗纳克中间体7-(对溴苯甲酰基)吲哚-2-酮的合成方法与流程
本发明涉及一种普罗纳克中间体的合成方法,更具体而言是涉及一种7-(对溴苯甲酰基)吲哚-2-酮(合成普罗纳克的关键中间体)的合成方法,属于有机化学合成领域。
背景技术:
普罗纳克(
7-(对溴苯甲酰基)吲哚-2-酮本身就是一种非常重要的含氮杂环化合物,可广泛用于药物合成中,例如其就是合成溴芬酸钠的关键中间体,可与naoh反应而得到溴芬酸钠(参见下面所列文献1-2),反应式如下:
然而,7-(对溴苯甲酰基)吲哚-2-酮的合成报道非常少,少数报道的方法主要如下:
us4126635和us4182774使用2-氨基-4’-溴-二苯甲酮为起始原料,通过四步反应制备得到了7-(对溴苯甲酰基)吲哚-2-酮:①在-70℃下,以2-氨基-4’-溴-二苯甲酮与2-甲巯基乙酸乙酯反应生成硫盐;②接着在三乙胺存在下发生重排反应生成3-取代2-氨基二苯甲酮;③在酸性条件下3-取代2-氨基二苯甲酮发生环合反应生成7-对溴苯甲酰基-3-(甲巯基)吲哚-2-酮;④在雷尼镍作用下,7-对溴苯甲酰基-3-(甲巯基)吲哚-2-酮被还原生成目标产物7-(对溴苯甲酰基)吲哚-2-酮,总收率小于30%,详细反应过程如下:
然而,上述合成方法使用了还需要多步反应才能制备得到的2-氨基-4’-溴-二苯甲酮作为起始原料,从而导致整体的反应路线过长,整体收率低,工艺繁琐。
ep0221753公开了一种7-(对溴苯甲酰基)吲哚-2-酮的制备方法,首先采用对溴苯甲腈和吲哚啉为原料,使用三氯化硼和三氯化铝为活化剂,进行friede-crafts酰化反应,生成7-(对溴苯甲酰基)吲哚啉;再经二氧化锰氧化反应生成7-(对溴苯甲酰基)吲哚;接着使用n-氯代丁二酰亚胺作为氯化试剂进行氯化反应,生成氯代7-(对溴苯甲酰基)吲哚;最后在磷酸存在下进行酸解反应,生成目标产物7-(对溴苯甲酰基)吲哚-2-酮,其反应式如下:
但该方法仍然需要四步反应才能实现7-(对溴苯甲酰基)吲哚-2-酮的合成,其总收率偏低,约为28%。此外,该方法的第一步反应使用了大量的对环境造成严重污染的三氯化铝作为活化剂。
夏泽宽等人(见下面所列文献3)对上述反应工艺(ep0221753)进行了改进:使用自制的活性二氧化锰作为氧化剂,其氧化产物7-(对溴苯甲酰基)吲哚的收率可以提高至92.4%(ep0221753中为85%);另外,还改变了氯化反应后处理方法,革除了乙醚,增加了二氯甲烷萃取,氯代7-(对溴苯甲酰基)吲哚的收率明显提高至97.9%(ep0221753中为64%)。整体而言,该改进方法虽然提高了7-(对溴苯甲酰基)吲哚-2-酮总收率,但其仍然非常低,约为33%。
除上述现有文献1-3外,国内外科学家还开发了类似化合物的、尤其是涉及氰基与硼酸基进行反应诸多合成方法,例如:
文献4中公开了芳基硼酸和腈类化合物通过镍催化而制备芳基酮的方法,其反应式如下:
但本发明人发现,当文献4采用邻亚氨基苯腈(例如本申请中的7-氰基吲哚-2-酮)作为反应原料时,按照文献4的方法仅能以很低产率得到7-(对溴苯甲酰基)吲哚-2-酮,针对如此缺陷,发明人认为可能有以下三方面的原因导致:
a、配位问题:由于亚氨基易与过渡金属催化剂配位导致催化剂失活,进而难以实现对氰基的加成反应;
b、电子效应问题:由于亚氨基属于较强的第一类定位基团,使得苯环的电子云密度增加,导致氰基的碳原子不易接受金属物种的加成,从而使反应难以进行。
综上所述,非甾体抗炎镇痛药普罗纳克关键中间体的现有合成工艺存在反应步骤繁琐、反应条件苛刻、收率过低等诸多问题,亟待解决,这对非甾体抗炎药镇痛普罗纳克的合成具有重要的理论和研究价值。
现有技术文献
文献1:welsteadjr.,w.j.;moran,h.w.;stauffer,h.f.;antiinflammatoryagents.1.synthesisandantiinflammatoryactivityof2-amino-3-benzoylphenylaceticacid[j].j.med.chem.1979,22,1074-1079。
文献2:walsh,d.a.;moran,h.w.;shamblee,d.a.;antiinflammatoryagents.3.synthesisandpharmacologicalevaluationof2-amino-3-benzoylphenylaceticacidandanalogues[j].j.med.chem.1984,27,1379-1388。
文献3:夏泽宽;于光恒;蔡霞;溴芬酸钠的合成[j].中国药科大学学报2003,34,405-406。
文献4:ying-chiehwongetal.,directsynthesisofarylketonesbynickel-catalyzedadditionofarylboronicacidstonitriles[j].organicletters,2010,12(8),1736-1739。
技术实现要素:
为了克服上述指出的缺陷,以及研发作为非甾体抗炎镇痛药普罗纳克关键中间体的7-(对溴苯甲酰基)吲哚-2-酮的合成方法,本发明人进行了大量的研究,在付出了创造性劳动后,从而完成了本发明。
需要指出的是,本发明是在“浙江省公益技术应用研究计划项目(项目编号:2016c31022)”的资助下完成的,在此谨表示感谢。
具体而言,本发明的技术方案和内容涉及一种普罗纳克中间体7-(对溴苯甲酰基)吲哚-2-酮的合成方法,所述方法包括:在镍催化剂和酸促进剂存在下,7-氰基吲哚-2-酮与对溴苯硼酸于溶剂中进行反应,从而得到7-(对溴苯甲酰基)吲哚-2-酮。
其反应式如下:
在本发明的所述合成方法中,所述镍催化剂选自ni(dppe)cl2、ni(acac)2、ni(dppf)cl2、ni(dppp)cl2、ni(pph3)2cl2、ni(pph3)2br2、nife2o4或nicl2中的任意一种,优选为ni(dppe)cl2或ni(dppp)cl2,最优选为ni(dppe)cl2。
在本发明的所述合成方法中,所述酸促进剂选自对甲苯磺酸一水合物、三氟乙酸、三氟甲磺酸、樟脑磺酸、甲磺酸、盐酸或乙酸中的任意一种,最优选为三氟甲磺酸。
在本发明的所述合成方法中,所述溶剂为四氢呋喃(thf)、水、甲苯、1,4-二氧六环、乙二醇二甲醚(dme)、二甲基甲酰胺(dmf)、2-甲基四氢呋喃(2-methf)、丙酮、甲醇或n-甲基吡咯烷酮(nmp)中的任意一种,最优选为2-甲基四氢呋喃(2-methf)。
所述溶剂的用量并没有特别的限定,只要能够使得反应顺利进行并方便后续操作即可,本领域技术人员可根据反应物的实际用量而选择合适的该溶剂用量,在此不再进行详细描述。
在本发明的所述合成方法中,反应温度为70-100℃,例如可为70℃、80℃、90℃或100℃。
在本发明的所述合成方法中,反应时间为12-24小时,例如可为12小时、15小时、18小时、21小时或24小时。
在本发明的所述合成方法中,7-氰基吲哚-2-酮与对溴苯硼酸的摩尔比为1:2-4,例如可为1:2、1:3或1:4。
在本发明的所述合成方法中,7-氰基吲哚-2-酮与镍催化剂的摩尔比为1:0.05-0.15,例如可为1:0.05、1:0.1或1:0.15。
在本发明的所述方法中,7-氰基吲哚-2-酮与酸促进剂的摩尔比为1:4-5,例如可为1:4、1:4.5或1:5。
在本发明的所述方法中,反应结束后可进行适当的后处理操作,例如:反应结束后,先将反应混合物旋转蒸发至干燥,然后加入足量乙酸乙酯使其溶解,之后加入适量的饱和碳酸氢钠水溶液和饱和氯化钠水溶液的混合水溶液进行洗涤,分离出有机层和水层,水层用乙酸乙酯萃取3次后,合并有机层(即合并混合水溶液洗涤后的有机层和乙酸乙酯萃取得到的有机层),用无水硫酸钠干燥,减压蒸馏,残留物通过快速柱色谱(以体积比1:2-3的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的7-(对溴苯甲酰基)吲哚-2-酮。
如上所述,本发明提供了一种作为非甾体抗炎镇痛药普罗纳克关键中间体的7-(对溴苯甲酰基)吲哚-2-酮的合成方法,所述方法通过合适反应体系的综合选择,从而克服了现有技术中存在的诸多缺陷,尤其是无法得到产物(即现有技术文献4)的严重缺陷,从而为普罗纳克的合成提供了易得原料,在该物质的医药合成领域具有显著的经济价值和应用潜力。
附图说明
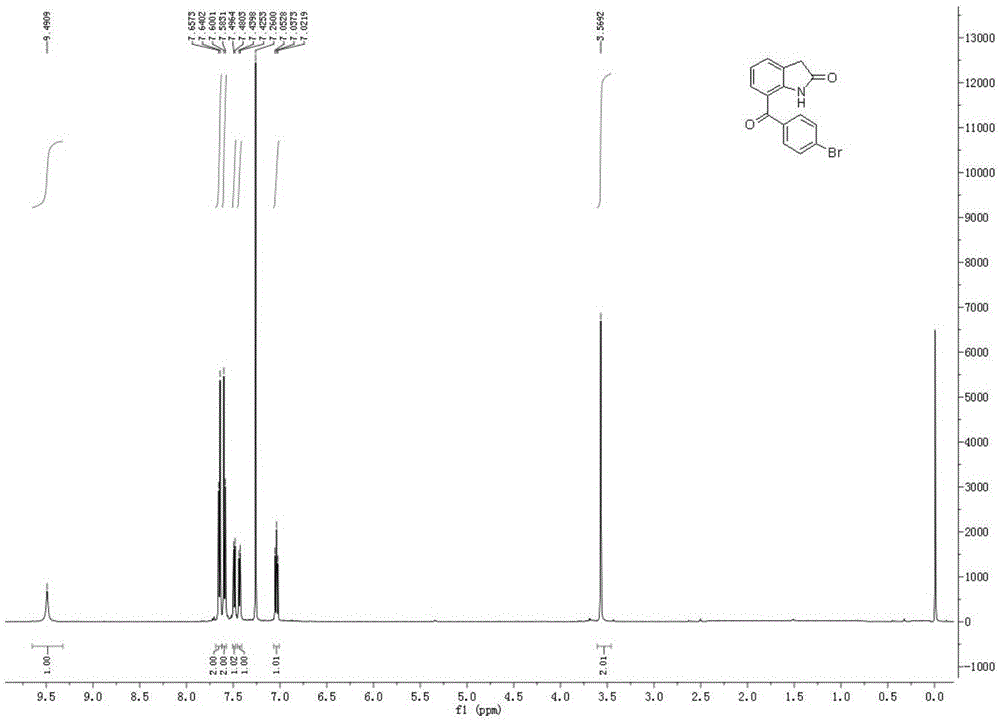
附图1是本发明方法所得7-(对溴苯甲酰基)吲哚-2-酮的h谱核磁共振谱图。
附图2是本发明方法所得7-(对溴苯甲酰基)吲哚-2-酮的c谱核磁共振谱图。
具体实施方式
下面通过具体实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
其中,在下面的所有实例中,反应路线为:
所得产物7-(对溴苯甲酰基)吲哚-2-酮的表征数据如下:
核磁:1hnmr(cdcl3,500mhz):δ9.49(s,1h),7.65(d,j=8.6hz,2h),7.59(d,j=8.5hz,2h),7.49(d,j=8.1hz,1h),7.43(d,j=7.3hz,1h),7.04(t,j=7.7hz,1h),3.57(s,2h)。
13cnmr(cdcl3,125mhz)δ195.6,176.4,145.7,136.7,131.7,130.9(2c),130.7(2c),129.2,127.2,121.1,117.6,112.0,35.1。
熔点:197-198℃。
实施例1
反应式如上,具体反应过程如下:
在室温下,向反应容器的适量溶剂(为2-甲基四氢呋喃)中加入100mmol7-氰基吲哚-2-酮、200mmol对溴苯硼酸、15mmol镍催化剂ni(dppe)cl2和400mmol三氟甲磺酸,然后搅拌升温至70℃,并在该温度下搅拌反应24小时。
反应结束后,先将反应混合物旋转蒸发至干燥,然后加入足量乙酸乙酯使其溶解,之后加入适量的饱和碳酸氢钠水溶液和饱和氯化钠水溶液的混合水溶液进行洗涤,分离出有机层和水层,水层用乙酸乙酯萃取3次后,合并有机层(即合并混合水溶液洗涤后的有机层和乙酸乙酯萃取得到的有机层),用无水硫酸钠干燥,减压蒸馏,残留物通过快速柱色谱(以体积比1:2的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的7-(对溴苯甲酰基)吲哚-2-酮,表征数据如上,产率为82.6%,纯度为95.9%。
实施例2
反应式如上,具体反应过程如下:
在室温下,向反应容器的适量混合溶剂(为2-甲基四氢呋喃)中加入100mmol7-氰基吲哚-2-酮、400mmol对溴苯硼酸、5mmol镍催化剂ni(dppe)cl2和500mmol三氟甲磺酸,然后搅拌升温至100℃,并在该温度下搅拌反应12小时。
反应结束后,先将反应混合物旋转蒸发至干燥,然后加入足量乙酸乙酯使其溶解,之后加入适量的饱和碳酸氢钠水溶液和饱和氯化钠水溶液的混合水溶液进行洗涤,分离出有机层和水层,水层用乙酸乙酯萃取3次后,合并有机层(即合并混合水溶液洗涤后的有机层和乙酸乙酯萃取得到的有机层),用无水硫酸钠干燥,减压蒸馏,残留物通过快速柱色谱(以体积比1:3的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的7-(对溴苯甲酰基)吲哚-2-酮,表征数据如上,产率为81.9%,纯度为95.3%。
实施例3
反应式如上,具体反应过程如下:
在室温下,向反应容器的适量混合溶剂(为2-甲基四氢呋喃)中加入100mmol7-氰基吲哚-2-酮、300mmol对溴苯硼酸、10mmol镍催化剂ni(dppe)cl2和450mmol三氟甲磺酸,然后搅拌升温至80℃,并在该温度下搅拌反应18小时。
反应结束后,先将反应混合物旋转蒸发至干燥,然后加入足量乙酸乙酯使其溶解,之后加入适量的饱和碳酸氢钠水溶液和饱和氯化钠水溶液的混合水溶液进行洗涤,分离出有机层和水层,水层用乙酸乙酯萃取3次后,合并有机层(即合并混合水溶液洗涤后的有机层和乙酸乙酯萃取得到的有机层),用无水硫酸钠干燥,减压蒸馏,残留物通过快速柱色谱(以体积比1:2.5的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的7-(对溴苯甲酰基)吲哚-2-酮,表征数据如上,产率为83.3%,纯度为94.8%。
实施例4
反应式如上,具体反应过程如下:
在室温下,向反应容器的适量混合溶剂(为2-甲基四氢呋喃)中加入100mmol7-氰基吲哚-2-酮、250mmol对溴苯硼酸、7.5mmol镍催化剂ni(dppe)cl2和475mmol三氟甲磺酸,然后搅拌升温至90℃,并在该温度下搅拌反应15小时。
反应结束后,先将反应混合物旋转蒸发至干燥,然后加入足量乙酸乙酯使其溶解,之后加入适量的饱和碳酸氢钠水溶液和饱和氯化钠水溶液的混合水溶液进行洗涤,分离出有机层和水层,水层用乙酸乙酯萃取3次后,合并有机层(即合并混合水溶液洗涤后的有机层和乙酸乙酯萃取得到的有机层),用无水硫酸钠干燥,减压蒸馏,残留物通过快速柱色谱(以体积比1:2.75的石油醚和乙酸乙酯的混合物作为洗脱剂)进行洗脱,收集洗脱液并蒸发除去洗脱剂,从而得到为淡黄色固体的7-(对溴苯甲酰基)吲哚-2-酮,表征数据如上,产率为82.7%,纯度为95.5%。
下面对该方法中的一些技术特征进行考察,从而对最优选条件进行了创造性选择,具体如下。
镍催化剂的考察
除分别将镍催化剂ni(dppe)cl2替换为下表1中的其它镍化合物外,其它操作均不变,从而重复实施了实施例1-4,所使用的镍催化剂、实施例对应关系和产物产率见下表1。
表1
其中,“nd”表示未检测到。由此可见,对于镍有机配体而言,优选为ni(dppe)cl2或ni(dppp)cl2,最优选为ni(dppe)cl2,当为其它镍催化剂则根本无法得到产物,这证明ni(dppe)cl2或ni(dppp)cl2可以良好产物得到目的产物,而ni(dppe)cl2则是最优选的催化剂。
酸促进剂的考察
除分别将三氟甲磺酸替换为下表2中的其它酸促进剂外,其它操作均不变,从而重复实施了实施例1-4,所使用的酸促进剂、实施例对应关系和产物产率见下表2。
表2
其中,“nd”表示未检测到。由此可见,对于酸促进剂而言,最优选三氟甲磺酸,当为其它算时或者无法得到产物,或者产率有大幅度的降低(甚至非常类似的甲磺酸也无法得到产物)。这证明三氟甲磺酸是最优选的酸促进剂。
省略三氟甲磺酸的考察
当将实施例1-4中的三氟甲磺酸均予以删除并替换为等量的zncl2时(即类似于现有技术文献4中的技术方案),则发现产率有大幅度降低(仅为19.2-25.7%)的产率,这证明三氟甲磺酸的存在对于该反应的顺利进行具有决定性的影响,其使用是必不可少的。
溶剂的考察
除将溶剂2-甲基四氢呋喃替换为下表3中的等量其它溶剂外,其它操作均不变,从而重复实施了实施例1-4,所使用的溶剂、实施例对应关系和产物产率见下表3。
表3
其中,“nd”表示未检测到。由此可见,对于溶剂而言,最优选2-methf,当为其它溶剂时或者无法得到产物,或者产率有大幅度的降低(即便是非常类似的thf,产率也大幅度降低为60.5%)。这证明2-methf是最优选的溶剂。
如上所述,本发明提供了一种作为非甾体抗炎镇痛药普罗纳克关键中间体的7-(对溴苯甲酰基)吲哚-2-酮的合成方法,所述方法通过合适反应体系的综合选择,从而克服了现有技术中存在的诸多缺陷,能够以高产率得到目的产物,从而为普罗纳克的合成提供了易得原料,在该物质的医药合成领域具有显著的经济价值和应用潜力。
应当理解,这些实施例的用途仅用于说明本发明而非意欲限制本发明的保护范围。此外,也应理解,在阅读了本发明的技术内容之后,本领域技术人员可以对本发明作各种改动、修改和/或变型,所有的这些等价形式同样落于本申请所附权利要求书所限定的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!