一种富马酸卢帕他定衍生物、其制备方法及中间体和用途与流程
本发明涉及药品制备领域,具体而言,涉及一种富马酸卢帕他定衍生物、其制备方法及中间体和用途。
背景技术:
富马酸卢帕他定作为一种组胺和血小板凝聚的双重拮抗剂,主要应用于治疗过敏性鼻炎和荨麻疹等过敏性疾病。富马酸卢帕他定自2003年3月以片剂方式首次在西班牙上市以来,至今已经英国、各欧盟成员国、美洲等全球多个国家上市,我国在2013年批准了国内制药企业生产富马酸卢帕他定原料药及制剂。
在药物的研究、生产、供应和临床使用过程中,必须保证药物的纯度,才能保证药物的有效和安全。药物中的杂质是影响其纯度的主要因素,一般而言,杂质是指药物中存在的无治疗作用或影响药物稳定性和疗效,甚至对人健康有害的物质。杂质来源大致可分为两类,一是由生产工艺和原辅料带入;二是在贮藏过程中受外界条件的影响,引起药物理化性质发生变化而产生。药品的不良反应除与活性成分自身的生理活性有关外,与药品中存在的杂质有密切的关系。所以规范地进行药品杂质的研究,并将杂质控制在一个安全、合理的限度范围之内,将直接关系到富马酸卢帕他定的质量和安全。
作为药品质量控制的一个重要环节,为了更好地控制药物质量,必须对杂质进行严格检测和监控。通过杂质的检测,弄清楚药品中杂质的来源、性质、检测方法及其限量,可以优化药品制造工艺,进而避免杂质的产生或将杂质降低到最低限度,从多方面保证和提高药品质量。对于富马酸卢帕他定杂质的制备方法研究意义重大,它可以用于富马酸卢帕他定原料药和制剂生产中的杂质定性及定量分析,从而可以提高富马酸卢帕他定原料药及制剂的质量标准,为人民群众的用药安全提供指导。
到目前为止,现有技术中针对富马酸卢帕他定的研究主要集中于产品本身的制备方法研究,而针对其生产过程中存在杂质的研究以及制备方法寥寥无几。申请人长期致力于研究富马酸卢帕他定的制备工艺以及杂质检测方法的优化,研究并申请了多件富马酸卢帕他定制备方法及其杂质专利,目前多件申请已获得授权。例如:
cn104098557a(申请日:2014.07.02)涉及一种富马酸卢帕他定杂质j及其制备方法,其采用富马酸卢帕他定(2)为原料,采用30%-50%的双氧水溶液反应得到富马酸卢帕他定杂质j粗品(1),粗品经过提纯得到杂质j纯品,纯度可达到98.5%。
cn108299395a(申请日:2017.12.08)涉及一种富马酸卢帕他定杂质s及其制备方法,杂质结构为
然而,现有技术中对于生产过程中所存在的杂质种类研究仍有待完善,仍有许多新的杂质未被发现,且如何满足杂质标准品的巨大合成需求,都有待进一步研究。
基于上述问题的考虑,本发明在生产工艺优化过程中发现了一种新的杂质富马酸卢帕他定衍生物,但是目前并没有合成该杂质的有效方法,使得该杂质的研究和控制成为一个难题。
技术实现要素:
本发明的主要目的在于提供一种富马酸卢帕他定衍生物、其制备方法及中间体和用途,以解决现有技术中富马酸卢帕他定的杂质富马酸卢帕他定衍生物无法有效合成导致其控制较为困难的问题。
为了实现上述目的,根据本发明的一个方面,提供了一种富马酸卢帕他定衍生物,富马酸卢帕他定衍生物具有以下结构式i或其药学上可接受的盐,
结构式i。
进一步地,上述制备方法包括:步骤s2,将化合物b与化合物c反应,制得富马酸卢帕他衍生物lp-4,其中,化合物b的结构式为
进一步地,上述化合物b和化合物c的摩尔比为1:0.5~1.4,优选为1:0.8。
进一步地,上述步骤s2的反应在缚酸剂存在下进行,缚酸剂为有机弱碱或无机弱碱,优选为三乙胺、吡啶、碳酸钾和碳酸钠组成的组中任意一种或多种,进一步优选化合物b与缚酸剂的摩尔比为1:1~5,优选为1:3;优选步骤s2的反应在第一溶剂中进行,第一溶剂为非质子性溶剂,更优选地,非质子性溶剂选自二氯甲烷和/或三氯甲烷;优选步骤s2的反应在室温或加热条件下进行,优选地,反应的温度为室温至50℃,更优选地,反应的温度为室温。
进一步地,上述制备方法还包括制备化合物b的步骤s1,步骤s1包括:使化合物a与氧化剂反应,制得化合物b,化合物a为5-甲-3-卤代甲基吡啶或5-甲-3-卤代甲基吡啶盐,优选5-甲-3-卤代甲基吡啶盐为5-甲-3-卤代甲基吡啶氢溴酸盐或5-甲-3-卤代甲基吡啶氢氯酸盐。
进一步地,上述化合物a与氧化剂的摩尔比为1:1~5,更优选为1:2。
进一步地,上述氧化剂选自间氯过氧化苯甲酸、mno2、双氧水、过氧乙酸、过氧乙酸盐和过氧苯甲酸中的任意一种;优选步骤s1的反应在催化剂条件下进行,催化剂选自碱和/或碱溶液,催化剂选自碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、三乙胺和各自的溶液中的任意一种,更进一步优选催化剂为碳酸氢钠水溶液;步骤s1的反应在第二溶剂中进行,第二溶剂为非质子性溶剂,更优选地,非质子性溶剂选自二氯甲烷和/或三氯甲烷;优选步骤s1的反应在室温或加热条件下进行,优选地,反应的温度为室温至50℃,更优选地,反应的温度为室温。
进一步地,上述卤原子为溴原子。
根据本发明的另一方面,提供了一种合成上述富马酸卢帕他定衍生物的中间体,中间体具有结构式
根据本发明的另一方面,提供了上述富马酸卢帕他定衍生物用作质量控制标准品的用途。
应用本发明的技术方案,本申请上述的富马酸卢帕他定衍生物为富马酸卢帕他定合成中的杂质,可以用于富马酸卢帕他定质量控制的标准品,为提高富马酸卢帕他定的纯度提供参考。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
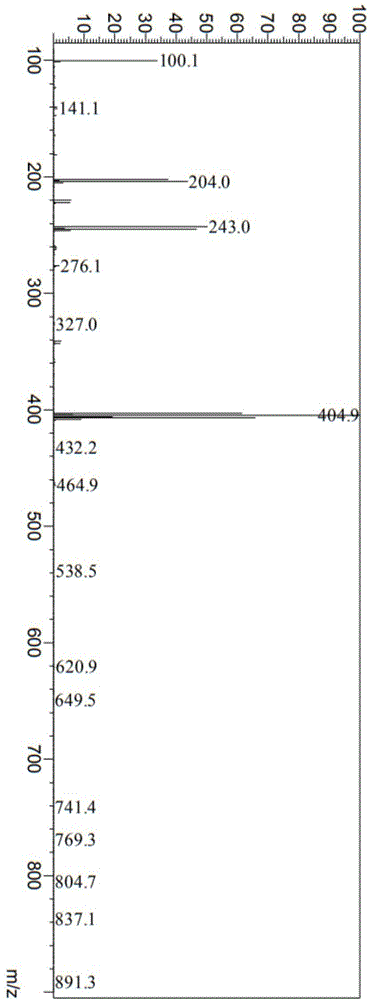
图1示出了实施例1得到的化合物b1-(2-羧基-1-羧乙基)-3-羟甲基-5-甲基吡啶-1-鎓的ms图;
图2示出了实施例2得到的富马酸卢帕他定衍生物lp-4的1h-nmr图;
图3示出了实施例2得到的富马酸卢帕他定衍生物lp-4的13c-nmr图;
图4示出了实施例2得到的富马酸卢帕他定衍生物lp-4的ms图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
如本申请背景技术所分析的,本申请在对富马酸卢帕他定的合成路线进行优化时,发现了新的富马酸卢帕他定衍生物,但是由于其含量太少,无法有效对其进行研究和控制,为了解决该问题,本申请提供了一种富马酸卢帕他定衍生物的制备方法及中间体和用途。
在本申请一种典型的实施方式中,提供了一种富马酸卢帕他定衍生物,该富马酸卢帕他定衍生物具有以下结构式i或其药学上可接受的盐,
结构式i。
本申请上述的富马酸卢帕他定衍生物(本申请称为富马酸卢帕他定衍生物lp-4)为富马酸卢帕他定合成中的杂质,可以用于富马酸卢帕他定质量控制的标准品,为提高富马酸卢帕他定的纯度提供参考。
在本申请另一种典型的实施方式中,提供了上述富马酸卢帕他定衍生物的制备方法,该制备方法包括:步骤s2,将化合物b与化合物c反应,制得富马酸卢帕他衍生物lp-4,其中,化合物b的结构式为
上述步骤s2的合成路线如下:
上述富马酸卢帕他定衍生物为富马酸卢帕他定衍生物lp-4,该制备方法采用较短的合成路线,能够提高富马酸卢帕他定衍生物lp-4的收率,同时该合成路线还具有副反应少,重现性好等优点。在此基础上,通过对富马酸卢帕他定衍生物lp-4的制备及结构鉴定,能够为富马酸卢帕他定的定性和定量分析提供品质优异、纯度较高的对照品,上述制备方法所制备的产品经提纯后纯度均大于99%,从而对富马酸卢帕他定的制备及其应用具有良好的指导意义。而且上述化合物d可以采用来源广、价格低廉的原料制备,因此降低了本申请制备方法实施的成本。
在一种实施例中,上述化合物b和化合物c的摩尔比为1:0.5~1.4,优选为1:0.8。通过上述摩尔比的控制保证各原料足够高的转化率。另外,为了进一步加速上述反应,优选步骤s2的反应在缚酸剂存在下进行,缚酸剂为有机弱碱或无机弱碱,优选为三乙胺、吡啶、碳酸钾和碳酸钠组成的组中任意一种或多种,进一步优选化合物b与缚酸剂的摩尔比为1:1~5,优选为1:3。另外,为了提高反应物的分散性进而提高反应效率,优选步骤s2的反应在第一溶剂中进行,第一溶剂为非质子性溶剂,更优选地,非质子性溶剂选自二氯甲烷和/或三氯甲烷。上述步骤s2的反应在室温或加热条件下均可进行,但是温度如果太低则反应进行缓慢甚至无法进行,如果温度太高则副产物增多,优选地,反应的温度为室温至50℃,更优选地,反应的温度为室温。
在本申请一种实施例中,上述制备方法还包括制备化合物b的步骤s1,上述步骤s1包括:使化合物a与氧化剂反应,制得化合物b,化合物a为5-甲-3-卤代甲基吡啶或5-甲-3-卤代甲基吡啶盐,优选5-甲-3-卤代甲基吡啶盐为5-甲-3-卤代甲基吡啶氢溴酸盐或5-甲-3-卤代甲基吡啶氢氯酸盐。
上述步骤s1中,当化合物a为5-甲-3-卤代甲基吡啶的反应路线为:
上述化合物a来源广泛,价格较低,且上述氧化反应的过程易于控制,因此使得化合物b的制备成本较低且收率较高。
为了提高物质的利用率,优选上述化合物a与氧化剂的摩尔比为1:1~5,更优选为1:2。
用于上述反应的氧化剂可以有多种,优选上述氧化剂选自间氯过氧化苯甲酸(m-cpba)、mno2、双氧水、过氧乙酸、过氧乙酸盐和过氧苯甲酸中的任意一种,以利于产物的分离。为了加快反应,优选步骤s1的反应在催化剂条件下进行,催化剂选自碱和/或碱溶液,催化剂选自碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、三乙胺和各自的溶液中的任意一种,更进一步优选催化剂为碳酸氢钠水溶液。另外,为了提高反应物的分散性进而提高反应效率,优选上述步骤s1的反应在第二溶剂中进行,第二溶剂为非质子性溶剂,更优选地,非质子性溶剂选自二氯甲烷和/或三氯甲烷。上述步骤s2的反应在室温或加热条件下均可进行,但是温度如果太低则反应进行缓慢甚至无法进行,如果温度太高则副产物增多,优选地,上述反应的温度为室温至50℃,更优选地,反应的温度为室温。
上述的卤原子可以选用常用的氯原子、溴原子或碘原子,优选上述卤原子为溴原子,以提高产物收率。
在本申请另一种典型的实施方式中,提供了一种合成上述富马酸卢帕他定衍生物的中间体,该中间体具有结构式
在本申请再一种典型的实施方式中,提供了一种上述富马酸卢帕他定衍生物用作质量控制标准品的用途。由于本申请的制备方法所得到的富马酸卢帕他定衍生物的纯度较高,因此可以用于富马酸卢帕他定的质量控制的标准品,为富马酸卢帕他定的制备具有良好的指导意义。
以下将结合实施例和对比例,进一步说明本申请的有益效果。
实施例1
在1l的三颈圆底烧瓶中通入惰性气氛n2并保持,加入3-溴甲基-5-甲基吡啶氢溴酸盐(化合物a,20g,74.92mmol),二氯甲烷(200ml)以及水(40g),接着在0℃下搅拌滴加碳酸氢钠(27.1g,322.58mmol)的水溶液(40ml)和m-cpba(37.1g,214.99mmol)的二氯甲烷溶液(200ml)。室温搅拌2小时,所得溶液用5x200ml二氯甲烷提取,合并有机层,用无水硫酸钠干燥,真空浓缩。残留物上硅胶柱用二氯甲烷/甲醇洗脱,得到7.5g(50%)化合物b3-溴甲烷-5-甲基吡啶-n-氧化物淡黄色固体,测试结果见图1。
产物的ms图谱数据为:
lc-ms-ph-jlu-lp-4-1:(es,m/z):202.0(m+1);204.0(m+1)。
实施例2
在250ml的圆底烧瓶中通入惰性气氛n2并保持,加入实施例1制得的化合物b(7.5g,37.12mmol),二氯甲烷(150ml)和三乙胺(11.2g,110.68mmol),在0℃下分批加入4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚烷[1,2-b]吡啶-11烯基)哌啶(化合物c,11.5g,37.00mmol)所得溶液在室温下搅拌过夜,接着在0℃下加入20ml水淬灭反应,分离有机层并真空浓缩。得到的粗产品采用flash-prep-hplc纯化,条件如下:色谱柱为c18填料;流动相,acn、水(0.1%nh4oh);梯度:20min内20%acn升至50%acn;检测器,uv254&220nm;粗产品进一步用prep-hplc纯化,条件如下:色谱柱为c18填料,19*150nm,流动相,acn和水(0.1%nh4oh);梯度:2min内38%acn,8min内38%-39%acn;检测器,uv254&220nm,得到3.05g(19%,纯度99.1%)富马酸卢帕他定衍生物lp-4粉色固体。对合成过程中获得的取代产物进行测试,测试结果见图2-4。
产物的1hnmr图谱、13cnmr及ms图谱数据为:
1h-nmr:(300mhz,dmso-d6,ppm)δ8.33(d,j=3.3,1h),8.01(s,1h),7.97(s,1h),7.57(d,j=6.9hz,1h),7.29(s,1h),7.22-7.16(m,2h),7.12(s,1h),7.06(d,j=8.4hz,1h),3.42(s,1h),3.32-3.23(m,2h),2.85-2.76(m,2h),2.64-2.59(m,2h),2.37-2.30(m,2h),2.23(s,3h),2.19-2.15(m,4h);
13cnmr(75mhz,dmso-d6,ppm)δ157.57,146.79,140.52,138.35,137.97,137.76,137.61,136.67,136.23,133.65,132.71,131.94,131.23,129.39,127.27,126.07,122.74,58.45,54.51,40.79,40.51,40.24,39.96,39.68,39.40,39.12,31.52,30.97,30.84,18.04;
lc-ms-ph-jlu-lp-4-0:(es,m/z):432.25(m+1)。
实施例3
与实施例1的区别为:化合物a和m-cpba的摩尔比为1:3,所得化合物b的收率为45wt%。
实施例4
与实施例1的区别为:化合物a和m-cpba的摩尔比为1:1,所得化合物b的收率为41wt%。
实施例5
与实施例1的区别为:化合物a和m-cpba的摩尔比为1:5,所得化合物b的收率为40wt%。
实施例6
与实施例1的区别为:将m-cpba替换为h2o2,所得化合物b的收率为40wt%。
实施例7
与实施例1的区别为:将m-cpba替换为mno2,所得化合物b的收率为45wt%。
实施例8
与实施例1的区别为:将碳酸氢钠替换为三乙胺,所得化合物b的收率为40wt%。
实施例9
与实施例1的区别为:化合物a替换为3-溴甲基-5甲基吡啶氢氯酸盐,所得化合物b的收率为48wt%。
实施例10
与实施例2的区别为:化合物b与三乙胺的摩尔比为1:2,所得产物的收率为12wt%。
实施例11
与实施例2的区别为:化合物b与三乙胺的摩尔比为1:1,所得产物的收率为10wt%。
实施例12
与实施例2的区别为:化合物b与三乙胺的摩尔比为1:5,所得产物的收率为20wt%。
实施例13
与实施例2的区别为:缚酸剂替换为碳酸钠,所得产物的收率为15wt%。
实施例14
与实施例2的区别为:化合物c与化合物b的摩尔比为0.8:1,所得产物的收率为25wt%。
实施例15
与实施例2的区别为:化合物c与化合物b的摩尔比为0.5:1,所得产物的收率为16wt%。
实施例16
与实施例2的区别为:化合物c与化合物b的摩尔比为1.4:1,所得产物的收率为21wt%。
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
本申请上述的富马酸卢帕他定衍生物为富马酸卢帕他定合成中的杂质,可以用于富马酸卢帕他定质量控制的标准品,为提高富马酸卢帕他定的纯度提供参考。
上述富马酸卢帕他定衍生物lp-4的制备方法采用较短的合成路线,能够提高富马酸卢帕他定衍生物lp-4的收率,同时该合成路线还具有副反应少,重现性好等优点。在此基础上,通过对富马酸卢帕他定衍生物lp-4的制备及结构鉴定,能够为富马酸卢帕他定的定性和定量分析提供品质优异、纯度较高的对照品,上述制备方法所制备的产品经提纯后纯度均大于99%,从而对富马酸卢帕他定的制备及其应用具有良好的指导意义。而且上述化合物d可以采用来源广、价格低廉的原料制备,因此降低了本申请制备方法实施的成本。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!