一种N-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺的合成方法与流程
本发明属于有机医药合成领域,具体涉及n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺的合成方法。
背景技术:
自二十世纪初发现抗生素以来,抗生素成了人类战胜病菌的神奇武器。然而,人们很快发现虽然新的抗生素层出不穷,但是,抗生素的耐药菌也越来越多,耐药菌的传播令人担忧。多重耐药(mdr)的联合选择(co-selection)的含义是对青霉素、头孢菌素类、氨基糖甙类、磺胺异唑类或喹诺酮等存在耐药。抗生素耐药性是目前科学家急需解决的难题,而2-氨基嘧啶类及嘧啶衍生物类药物对细菌的耐药性有很大的作用。本发明的n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺是氨基嘧啶类的重要中间体。该路线反应类型简单,操作简单环保,适合于工艺放大。
技术实现要素:
发明目的:本发明的目的在于提供一种合成n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺的方法。本发明所解决的技术问题是,合成了一种新型嘧啶类中间体,其合成路线简单,可操作性强,适合工艺放大。
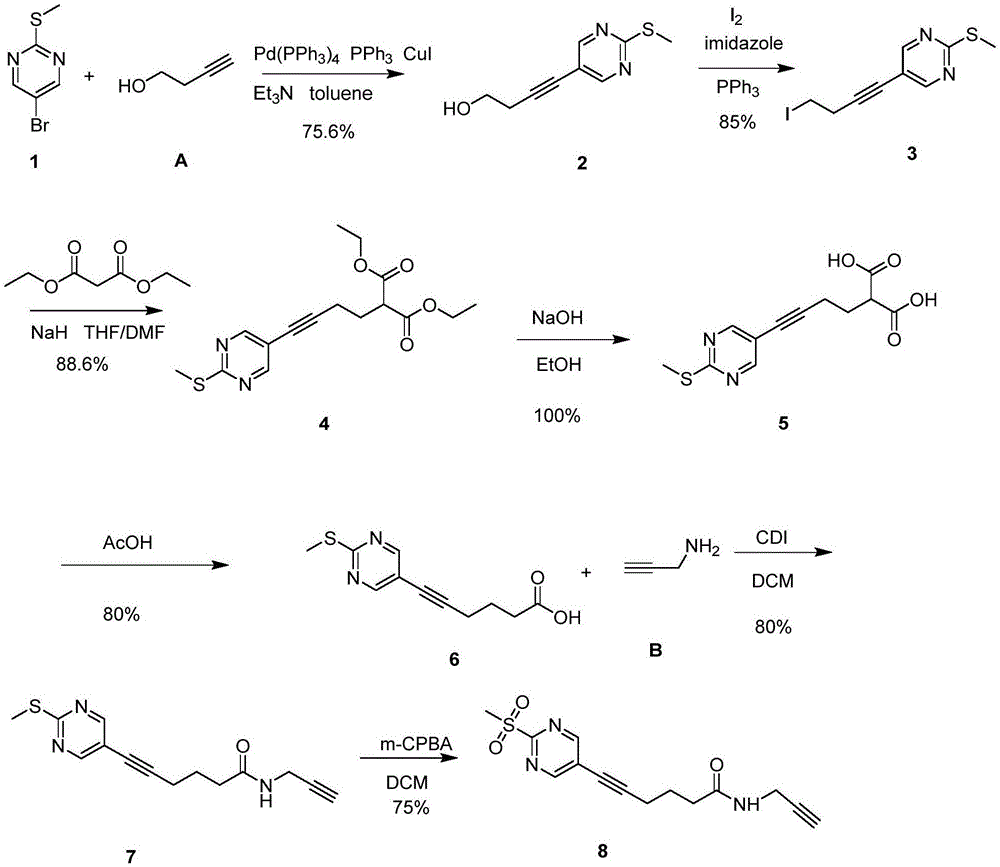
技术方案:本发明n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺的合成反应路线为:
其中,化合物1为5-溴-2-甲巯基嘧啶;化合物a为3-丁炔-1-醇;化合物2为4-(2-(甲基硫基)嘧啶)-3-丁炔-1-醇;化合物3为4-(2-(甲硫基)嘧啶)-3-丁炔-1-碘代物;化合物4为4-(2-(甲基硫基)嘧啶)-3-丁炔-1-丙二酸二乙酯;化合物5为4-(2-(甲巯基)嘧啶)-3-丁炔-1-丙二酸;化合物6为4-(2-(甲硫基)嘧啶)-5-己炔酸;化合物7为n-(2-丙炔)-6-(2-(甲基硫基)嘧啶)-5-己炔酰胺;化合物b为炔丙胺,目标物化合物8为n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺。
步骤包括:(1)5-溴-2-甲巯基嘧啶与3-丁炔-1-醇通过sonogashira偶联反应得到4-(2-(甲巯基)嘧啶)-3-丁炔-1-醇;
(2)通过appel反应将4-(2-(甲硫基)嘧啶)-3-丁炔-1-醇生成4-(2-(甲基硫基)嘧啶-3-丁炔-1-碘代物;
(3)4-(2-(甲基硫基)嘧啶)-3-丁炔-1-碘代物与丙二酸二乙酯生成4-(2-(甲硫基)嘧啶)-3-丁炔-1-丙二酸二乙酯;
(4)丙二酸二乙酯生成4-(2-(甲硫基)嘧啶)-3-丁炔-1-丙二酸二乙酯碱水解得到4-(2-(甲硫基)嘧啶-3-丁炔-1-丙二酸;
(5)4-(2-(甲硫基)嘧啶-3-丁炔-1-丙二酸脱羧生成4-(2-(甲硫基)嘧啶)-5-己炔酸;
(6)4-(2-(甲硫基)嘧啶-5-己炔酸与炔丙胺缩合生成n-(2-丙炔)-6-(2-(甲硫基)嘧啶)-5-己炔酰胺;
(7)n-(2-丙炔)-6-(2-(甲硫基)嘧啶-5-己炔酰胺氧化生成目标产物n-(2-丙炔)-6-(2-(甲磺酰基)嘧啶)-5-己炔酰胺。
步骤(1)具体为:取化合物1、化合物a、碱、铜盐与溶剂混合,在氮气气氛或惰性气体气氛下加入催化剂及配体至反应完全,过滤取滤液浓缩得到化合物2。其中碱为叔丁醇钠、叔丁醇钾、碳酸钾、磷酸钾或三乙胺,更优选三乙胺;
溶剂优选为甲苯、四氢呋喃、二氧六环,更优选为甲苯;
催化剂优选pdcl2、pdcl2(dppf)、pd(oac)2、pd(pph3)4、pd(pph3)2cl2和nicl2(dppf),更优选pd(pph3)4;
配体优选三苯基膦、2-二环己基磷-2',4',6'-三异丙基联苯(xphos)、三环己基膦等,更优选三苯基膦;
铜盐包括cui、cubr等,优选为cui;
化合物1、化合物a、碱、cui、催化剂及配体的投料摩尔比为1:1.0~3.0:1.0~5.0:0.01~0.5:0.01~0.05:0.01~0.5,更优选比例为1.0:2.0:3.0:0.05:0.015:0.03。
步骤(2)具体为:取化合物2、碱与有机溶剂混合,-10~10度下加入三苯氧膦(pph3)后反应1分钟~1.0小时,然后-10~10度下,加入碘源室温反应0.1~48小时,优选为0.5~24小时,tlc(pe/ea=2/1)检测反应完全,过滤,萃取纯化,浓缩结晶得到化合物3。
其中,碱为吡啶、三乙胺或咪唑,更优选咪唑。碱与化合物2摩尔比为1.2~5.0:1,更优选3.0:1.0。
进一步的,有机溶剂为二氯甲烷(dcm)或乙腈,以能充分搅拌为准,优选为8v。
碘源优选为n-碘代丁二酰亚胺(nis)或碘单质,更优选碘单质。碘源与化合物2摩尔比为:1.0~3.0:1。更优选为2.0:1。
加入三苯基膦后充分反应至生成中间体再加入碘源,加入碘源后反应至中间体充分生产产物。后处理浓缩后需要纯化处理掉的主要副产物为三苯氧膦,纯化所需要的溶剂包括但不限于常用的正己烷、甲基叔丁基醚(mtbe)、乙酸乙酯(ea)、二氯甲烷(dcm)、乙醇或甲醇,优选乙醇。
步骤(3)具体为:取碱与有机溶剂混合,冷至-10~10度,滴加丙二酸二乙酯,滴完后,室温搅拌反应0.1分钟~2.0小时(优选为0.5小时),然后加入化合物3,室温搅拌反应至原料反应完;倒入冰水中,萃取浓缩得黄色固体化合物4。
其中,碱为氢氧化钠、碳酸钾、钠氢或甲醇钠,更优选钠氢。丙二酸二乙酯、碱与化合物3的投料比为2.0~6.0:1.0~3.0:1,优选为4.0:3.0:1。
步骤(3)选用的溶剂为四氢呋喃、乙醇、甲醇、异丙醇、二氧六环、dmf和nmp中一种或混合溶剂,更优选体积比dmf/thf=1/3的混合溶剂。更进一步的,溶剂与化合物3的体积比优选5~20v,更优选的为15v。
优选的与化合物3的投料摩尔比为4.0:1,更优选的反应时间为0.5小时。
加入化合物3后反应时间优选为0.1~48小时,更优选为24小时。
反应结束后用甲基叔丁基醚或乙酸乙酯萃取,合并有机相,洗涤干燥并浓缩得到化合物4。
步骤(4)具体为:取化合物4与有机溶剂混合,加入碱搅拌反应至反应完全,浓缩干燥,得到化合物5。
步骤(4)所述的碱与化合物4的摩尔比为1~10:1,更优选为6:1。所述的碱为氢氧化钠、碳酸钾、钠氢或甲醇钠,优选为氢氧化钠。
步骤(4)所使用的溶剂为四氢呋喃、乙醇、甲醇、异丙醇、二氧六环、dmf或nmp,更优选乙醇。
步骤(4)的反应时间为0~48小时,优选为0.5~24小时,在本发明的一个优选方案中,反应时间为15小时。
步骤(5)具体为,化合物5与脱羧试剂反应1分钟~36小时,优选为0.5~24小时,至反应完全,萃取浓缩得到化合物6。
其中,脱羧试剂选自乙酸、二甲亚砜与氯化钠或氯化锂的混合物、氢氧化钠,氢溴酸、盐酸,更优选为乙酸。
步骤(6)具体为:化合物6与有机溶剂混合后加入缩合剂至反应完全;再加入化合物b(炔丙胺)反应至完全;萃取去除溶剂,得到化合物7。
其中,步骤(6)的有机溶剂选自二氯甲烷、氯仿、四氢呋喃或dmf,优选二氯甲烷。更进一步,溶剂体积优选为2.0v~10.0v。
缩合剂优选dcc,cdi,bop,hobt,edci,更优选cdi。化合物6、缩合剂与炔丙胺的摩尔比为1:1.0~6.0:1.0~6.0,更优选为1:5.0:5.0。加完cdi后反应时间以充分转换为中间体为准,在0~10.0小时之间,更优选2.0小时。加完炔丙胺后反应时间以充分生成产品为准,0~24小时之间,更优选1.0小时。
步骤(7)具体为:化合物7与有机溶剂混合,加入氧化剂后反应0.5~48小时至反应完全,洗涤后去除溶剂,纯化得到化合物8。
其中有机溶剂为二氯甲烷(dcm)、四氢呋喃(thf)、二甲基甲酰胺(dmf)、乙酸乙酯(ea)、甲醇或乙醇,优选为dcm。
所述的氧化剂为间氯过氧苯甲酸(m-cpba)、叔丁基过氧化氢(tbhp)、过氧化二苯甲酰(bpo)、二氧化锰或双氧水等,更优选m-cpba。氧化剂与化合物7的摩尔比为1~6:1,更优选为4.0:1。
纯化所使用的试剂包括但不限于正己烷、正庚烷、二氯甲烷、异丙醇、甲醇、乙醇、n-甲基吡咯烷酮(nmp)等常用溶剂。
本发明提供了一条简便有效的合成路线,原料市场易得,反应选择性高、反应温和、后处理简单(无需柱层析)的优点。此路线收率高,污染及危险性均较小,为工业生产提供了可能。
附图说明
图1为本发明的合成路径。
图2为化合物2的核磁图谱。
图3为化合物8的核磁图谱。
图4为实施例4化合物8的hplc图谱。
具体实施方式
下面结合具体实施实例对本发明进一步说明,具体实施例的描述本质上仅仅是范例,以下实施实例基于本发明技术方案中的合成路线进行实施,所用的原料和实际均为市售产品。具体合成路径如图1所示。
实施例14-(2-(甲巯基)嘧啶)-3-丁炔-1-醇的制备
取化合物1(103g,0.5022mol,1.0eq),化合物a(70.4g,1.0045mol,2.0eq),三乙胺(152.5g,1.5067mol,3.0eq),cui(4.8g,0.0251mol,0.05eq)和甲苯500ml加入到反应瓶中,氮气置换两次,然后加入pd(pph3)4(8.7g,0.0075mol,0.015eq)、pph3(4.0g,0.0151mol,0.03eq),氮气保护下升温到50度反应16个小时,tlc检测反应(pe/ea=2/1),原料基本上反应完毕。停止加热,冷却,硅藻土垫滤,滤液浓缩得淡黄色固体化合物2(73.7g,0.3794mol),产率75.6%。
产品核磁如图2:1hnmr(600mhz,dmso)δ8.67(s,2h),4.95(s,1h),3.60(m,2h),2.61(t,j=6.7hz,2h),2.51(d,j=10.1hz,3h),ms(m+1):195。
实施例24-(2-(甲巯基)嘧啶)-3-丁炔-1-碘代物的制备
化合物2(756g,3.892mol,1.0eq),咪唑(794g,11.676mol,3.0eq),加入到dcm6000ml中,机械搅拌,冷却到10度以下,分次加入pph3(2047g,7.784mol,2.0eq)反应0.5个小时,然后在零以下,分次加入i2(1977g,7.784mol,2.0eq),然后室温反应两个小时,tlc检测反应(pe/ea=2/1),原料反应完毕。垫硅藻土过滤,dcm淋洗,加水,分层,dcm反洗一次,合并dcm,盐水洗一次,干燥,浓缩,乙醇结晶得白色固体,产率85%。
采用1hnmr(600mhz,cdcl3)对制备的4-(2-(甲巯基)嘧啶)-3-丁炔-1-碘代物进行结构确认,1hnmr图谱化学位移为δ8.52(s,2h),3.51-3.54(m,2h),3.03-3.01(m,2h),2.57(s,3h),碘代物ms(m+1):305。
实施例34-(2-(甲巯基)嘧啶)-3-丁炔-1-丙二酸二乙酯的制备
取nah(60%,32.7g,0.8166mol,3.0eq)加入到dmf/thf=1/3的混合溶液1000ml中,搅拌,冷却到零度以下,滴加丙二酸二乙酯(174.5g,1.089mol,4.0eq),滴完后,室温搅拌30min。然后加入化合物3(70g,0.2722mol,1.0eq),室温搅拌反应24小时。tlc检测原料反应完。倒入冰水中,甲基叔丁基醚萃取三次,合并有机相,盐水洗一次,干燥,然后浓缩纯化得黄色固体化合物4(81.1g,0.2412mol),产率88.6%。
采用1hnmr(600mhz,cdcl3)对制备的4-(2-(甲基硫基)嘧啶)-3-丁炔-1-丙二酸二乙酯进行纯度确认,1hnmr图谱化学位移为δ8.39(s,2h),4.23-4.06(m,4h),3.52-3.50(m,1h),2.74-2.70(m,2h),2.57(s,3h),1.30-1.20(m,8h),产物ms(m+1):337。
实施例44-(2-(甲巯基)嘧啶)-3-丁炔-1-丙二酸二酸的制备
取化合物4(142.6g,0.4239mol,1.0eq),加入到700ml无水乙醇中搅拌,然后加入naoh(100g,2.5mol,6.0eq)室温搅拌反应15小时。tlc检测都反应生成化合物5。停止反应,冰浴下加入乙酸,搅拌,旋干,甲苯带干直接用于下步投料。
采用1hnmr(600mhz,cdcl3)鉴别,1hnmr(600mhz,cdcl3)δ8.87(s,2h),3.52-3.50(m,1h),2.74-2.70(m,2h),2.57(s,3h),1.95-1.75(m,2h),产物ms(m+1):281。
实施例54-(2-(甲巯基)嘧啶)-5-己炔酸的制备
将上步所得产品加入乙酸700ml,升温回流反应16小时。检测化合物4反应完毕,旋干,甲苯带一次溶剂,然后加入水和ea溶解,ea萃取三次,合并ea,盐水洗一次,干燥,旋干,得粗品黄色固体化合物6(80g,340mmol),两步产率80%。
1hnmr(600mhz,cdcl3)δ8.87(s,2h),2.74-2.70(m,2h),2.57(s,3h),2.50-2.30(m,2h),1.95-1.75(m,2h),产物ms(m+1):237。
实施例6n-(2-丙炔)-6-(2-(甲巯基)嘧啶)-5-己炔酰胺的制备
取化合物6(49.3g,0.2078mol,1.0eq),加入到500mldcm中搅拌,然后分次加入cdi(168.5g,1.039mol,5.0eq),加完后室温搅拌反应2小时。然后再加入炔丙胺(57g,1.039mol,5.0eq),加完,室温搅拌反应1小时,lcms检测都反应完毕。停止反应,加入冰水,分层,水用dcm萃取两次,合并有机相,用酸水洗一次,盐水洗一次,干燥,旋干,得粗品黄色固体化合物7(43.2g,0.1582mol),产率76.1%。
1hnmr(600mhz,cdcl3)δ8.87(s,2h),6.20(s,1h),4.07-4.05(m,2h),3.38-3.36(m,3h),2.61-2.59(m,2h),2.42-2.40(m,2h),2.24-2.23(m,1h),2.05-1.99(m,2h),产物ms(m+1):274。
实施例7n-(2-丙炔)-6-(2-(甲基磺酰基)嘧啶)-5-己炔酰胺的制备
取化合物7(43.2g,0.1582mol,1.0eq),加入到500mldcm中搅拌,然后分次加入mcpba(108.9g,0.6326mol,4.0eq),加完后室温搅拌反应2小时。tlc检测反应(pe/ea=2/1),原料反应完毕,停止反应,加入饱和碳酸氢钠溶液,分层,dcm层用饱和碳酸氢钠溶液再洗两次,盐水洗一次,干燥,旋干,dcm与正己烷混合溶剂(体积比1:5)结晶得白色固体化合物8(36.2g,118.6mmol,纯度99%,单杂小于0.5%),产率75%,hplc图谱如图4。
1hnmr(600mhz,cdcl3)δ8.86(s,2h),5.92(s,1h),4.07(dd,j=5.3,2.5hz,2h),3.36(s,3h),2.60(t,j=7.0hz,2h),2.40(t,j=7.3hz,2h),2.24(t,j=2.5hz,1h),2.02(p,j=7.1hz,2h),
产物ms(m+1):306,核磁如图3。
以上实施例只是对本发明的技术构思起到说明示例作用,并不能以此限制本发明的保护范围,本领域技术人员在不脱离本发明技术方案的精神和范围内,进行修改和等同替换,均应落在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!