一种磷丙泊酚钠的制备方法与流程
本发明涉及医药领域,特别是一种磷丙泊酚钠的制备方法。
背景技术:
磷丙泊酚钠(fospropofoldisodium)是2008年12月12日由美国食品药品管理局(fda)批准上市,临床上用作成年患者进行诊断或治疗操作过程的镇静催眠剂,该药为静脉注射剂。磷丙泊酚钠是丙泊酚的一种水溶性的前体药物,该前药静脉注射给药后在体内被内皮细胞表面的碱性磷酸酶代谢产生活性药物丙泊酚,其在大脑组织内迅速达到平衡,从而发挥剂量依赖性的镇静催眠作用。
其现有的合成方法有:专利wo0008033采用丙泊酚与氯甲硫醚反应,然后与氯化亚砜反应,再与苄基保护了磷酸银盐反应,最后脱苄基得到产物,所用金属盐催化剂价格昂贵,所用试剂污染严重,后处理环保压力很大,且收率只有72%,成本较高。该专利所述的其他方法收率更低。
专利us6204257采用丙泊酚为起始原料,经过六步反应得到磷丙泊酚钠,制备步骤较多,而且制备过程使用了价格昂贵且具有强致癌性的试剂,对环境和人体伤害都较大,不适合工业化生产。
cn101367834和cn102382133合成步骤虽然简单,但合成过程中氯溴甲烷或碘溴甲烷所使用的量为丙泊酚摩尔量的至少10倍甚至30倍量,磷酸的用量不少于至少3倍甚至10倍量。导致反应结束后产生大量的含卤素废液和废磷酸,难以处理,三废排放量大,环保压力大,亦不适合工业化生产。因此,磷丙泊酚钠的制备方法的改进和创造是目前亟需解决的问题。
技术实现要素:
针对上述情况,为解决现有技术之缺陷,本发明之目的就是提供一种磷丙泊酚钠的制备方法,可有效解决现有合成方法收率低,污染严重,难以工业化的问题。
本发明解决的技术方案是,反应结构式为:
具体包括以下步骤:
i)制备中间体3:将丙泊酚1、卤代甲酸酯2、碱性催化剂、有机溶剂a混合,在0~25℃反应2~4小时,反应结束后加丙泊酚1.5~2倍的水搅拌淬灭反应,搅拌状态下用2m的盐酸调ph到7,静置分层,收集有机层,有机层用无水硫酸镁干燥3~4小时,45~55℃减压蒸除溶剂,得粘稠物,粘稠物用丙泊酚0.1~0.3倍的乙酸乙酯分散,然后搅拌下缓慢滴加石油醚,石油醚用量为丙泊酚重量的2倍,析晶得固体中间体3;
所述的卤代甲酸酯为氯代甲酸酯或溴代甲酸酯,氯代甲酸酯或溴代甲酸酯中r烃基团为甲基、乙基、苯基、苄基中的一种;卤代甲酸酯的用量为丙泊酚的1.0~3.0倍,优选1.1倍;
所述的碱性催化剂为醇钠、氢氧化钠、氢氧化钾中的一种或三乙胺、乙二胺、n,n-二异丙基乙胺、1,8-二氮杂二环十一碳-7-烯中的一种;其用量为丙泊酚摩尔量的1.0~3.0倍,优选1.2倍;
所述的有机溶剂a为1,2-二氯乙烷、乙二醇二甲醚、氯仿、二氯甲烷中的一种,用量为丙泊酚重量的2~5倍;
ii)制备中间体4:将中间体3加入到有机溶剂b中,加入还原剂,在5~80℃反应0.5~4小时,反应结束后加中间体3重量的0.07-0.08倍的丙酮淬灭反应,加入中间体3重量的0.3-0.4倍的饱和碳酸钾水溶液搅拌均匀后静置分层,取上层清液经浓缩回收溶剂得中间体4;
所述的还原剂为硼氢化钠、硼氢化钾、硼氢化钠、氯化钙摩尔比2:1的混合物、硼氢化钠、氯化锌摩尔比2:1的混合物中的一种;还原剂用量为中间体3摩尔量的0.5~2.0倍,优选1.0倍;
所述的有机溶剂b为甲醇、乙醇、四氢呋喃,二氧六环,二氧五环,异丙醚中的一种,用量为中间体3重量的2~5倍;
iii)制备磷丙泊酚钠5粗品:中间体4溶解在有机溶剂c中,将磷酸化试剂加入到反应体系中,加热回流分水,反应2~4小时,降温,向体系加入与有机溶剂c等体积的水,浓盐酸调ph值到1~2,分层后有机层经干燥、浓缩回收有机溶剂c,得粘稠残留物,所得残留物用3~5倍重量的无水乙醇分散均匀,然后加入钠盐或其饱和水溶液反应30分钟,析出白色结晶为磷丙泊酚钠5;
所述的磷酸化试剂为焦磷酸,其用量为中间体4摩尔量的0.5~2.0倍,优选0.7倍。
所述的钠盐为氢氧化钠、碳酸钠或异辛酸钠中的一种,其用量为中间体4摩尔量的2.0~4.0倍,优选2.2倍。
所述的有机溶剂c为甲苯,二甲苯,环己烷,正己烷,石油醚中的一种,用量为中间体4重量的2~5倍。
iiii)磷丙泊酚钠5的纯化:取步骤iii)中制备的磷丙泊酚钠粗品,加入磷丙泊酚钠粗品重量6~8倍量的丙酮,在45~55℃溶解,待溶液溶清后,加入磷丙泊酚钠粗品0.005~0.01倍重量的药用活性炭回流30分钟,降温到50℃,过滤,滤液先搅拌下恢复到18~25℃,然后用水浴降温到18~22℃,保持30分钟,最后用冰水降温到8~12℃,保持2小时,析出白色结晶,过滤,即得白色固体,用预冷丙酮洗涤白色固体,所得固体在40℃减压干燥12小时,得类白色固体,即纯化磷丙泊酚钠5。
本发明生产工艺简单,中间体无需纯化即可用于下一步反应;大幅提高了总收率(总收率达91%),显著降低了成本;有机溶剂易回收利用,三废少且处理简单,环保友好,易于工业化,是磷丙泊酚钠的制备方法上的创新。
附图说明
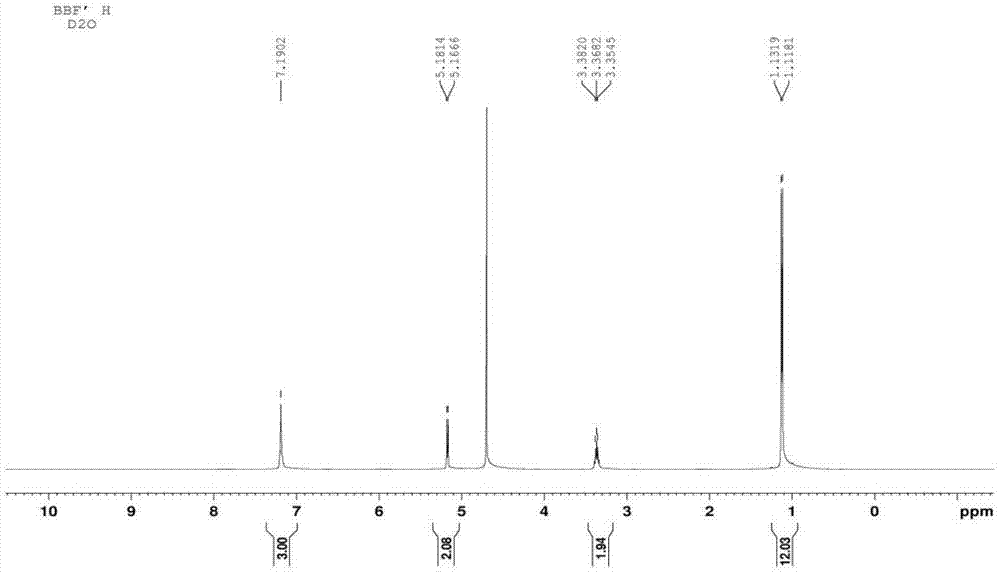
图1为本发明制备的磷丙泊酚钠的核磁共振氢谱。
图2为本发明制备的磷丙泊酚钠的核磁共振碳谱。
具体实施方式
以下结合附图和实施例对本发明的具体实施方式作进一步详细说明。
实施例1
i)在5l反应瓶中加入丙泊酚500g,1,2-二氯乙烷1l、氯甲酸甲酯290g、n,n-二异丙基乙胺434g,搅拌,在0~5℃反应2小时,薄层色谱检测至反应结束,向反应瓶中加入800ml纯水,搅拌状态下用2m的盐酸调ph到7,静置分层,收集有机层,加入无水硫酸镁干燥4小时,然后50℃减压回收1,2二氯乙烷,得淡黄色油状粘稠物,粘稠物用100ml乙酸乙酯分散,然后搅拌下缓慢滴加石油醚1000ml,析出类白色固体(中间体3)650.6克,收率98.3%。
ii)将上述类白色固体650克加入到5l反应瓶中,加入无水乙醇1l,硼氢化钠157克,25℃反应一小时后再加热回流2小时,液相检测反应至结束,加入50ml丙酮搅拌十分钟淬灭反应,加入饱和的碳酸钾溶液200ml,充分搅拌后静置分层,汲取上层清液,浓缩至干,得到粘稠物(中间体4)568.7克,收率99.2%。
iii)向步骤ii中所得粘稠物568克中加入600ml环己烷,搅拌分散均匀,搅拌状态下,缓慢滴加焦磷酸389克,加完后升温至回流分水器分水,至不再有水生成时停止加热,降至18-25℃后加入600ml水,用浓盐酸调ph到1~2,分层后,收集有机层用无水硫酸镁干燥,干燥结束后减压浓缩回收环己烷,得粘稠残留物,然后向残留物中加入无水乙醇1000ml分散,加入饱和氢氧化钠水溶液462克,析出大量固体,过滤、洗涤,烘干得白色结晶(磷丙泊酚钠)887.8克,收率98.0%。
实施例2:磷丙泊酚钠的制备
i)在2l反应瓶中加入丙泊酚200g,1,2-二氯乙烷1l、氯甲酸甲酯116g、三乙胺136g,搅拌,控温在0~5℃反应2小时,薄层色谱检测反应结束,向反应瓶中加入300ml纯水,搅拌状态下用2m的盐酸调ph到7,静置分层后,收集有机层,加入无水硫酸镁干燥4小时,然后50℃减压蒸除溶剂,得淡黄色油状粘稠物,粘稠物用30ml乙酸乙酯分散,然后滴加环己烷400ml,析出类白色固体(中间体3)260克,收率98.5%;
ii)将上述类白色固体260克加入到2l反应瓶中,加入无水四氢呋喃800ml,硼氢化钠63克,氯化钙90克,20℃搅拌30分钟,降温到10℃,将上述260克类白色固体的四氢呋喃溶液滴加到反应瓶中,加完后回流,液相检测至反应结束,降温0℃,缓慢加入丙酮20ml搅拌十分钟淬灭反应,加入饱和的碳酸钾溶液90ml,充分搅拌后静置分层,汲取上层清液,浓缩至干,得到粘稠物(中间体4)226.7克,收率98.9%;
iii)将步骤ii中所得粘稠物226克用250ml甲苯分散,搅拌状态下,缓慢滴加焦磷酸136克,加完后升温至回流,分水,至不再有水生成时降温到18-25℃,加入250ml水,用浓盐酸调ph到1~2,分层后,收集有机层用无水硫酸镁干燥,干燥结束后减压浓缩回收甲苯,然后向残留物中加入无水乙醇500ml分散,加入异辛酸钠462克,充分搅拌后,析出大量固体,抽滤,洗涤,烘干得白色结晶(磷丙泊酚钠5)356克,收率98.6%。
实施例3:磷丙泊酚钠的精制
取磷丙泊酚钠粗品100g,用丙酮800ml,在50℃溶解,待溶液溶清后,加入0.5g药用活性炭回流30分钟,降温到50℃,过滤,滤液先搅拌下恢复到18-25℃,然后用水浴降温到20℃保持30分钟,最后用冰水降温到10℃保持2小时析出白色结晶,过滤即得白色固体,用预冷丙酮洗涤白色固体,所得固体在40℃减压干燥12小时,得类白色固体(纯化磷丙泊酚钠5)95.2g。实验数据如下:纯度99.6%,ir:
经结构分析确证,本方法所制备的磷丙泊酚钠与文献报道的完全一致,同样必定具有相同的药理活性。核磁共振氢谱、碳谱如图1-2所示。
本发明是将丙泊酚在碱的催化作用下和卤代甲酸酯生成2,6-二异丙基苯氧基甲酸酯(中间体3);2,6-二异丙基苯氧基甲酸酯经还原得到2,6-二异丙基苯氧基甲醇(中间体4);2,6-二异丙基苯氧基甲醇再与焦磷酸酯化,然后与钠盐作用生成磷丙泊酚钠(5)。经纯化,得到高纯度磷丙泊酚钠。该生产工艺简单,中间体无需纯化即可用于下一步反应;大幅提高了总收率(总收率达91%),显著降低了成本;有机溶剂易回收利用,三废少且处理简单,环保友好,易于工业化。
本发明方法简单,易操作,工艺中间体无需纯化即可用于下一步反应;原料来源稳定且廉价易得,成本低,产品质量好,收率高(精制后总收率达91%);有机溶剂易回收利用,三废少且处理简单,环保友好,易于工业化,经济和社会效益巨大。
- 还没有人留言评论。精彩留言会获得点赞!