一种阿奇霉素重排杂质内酰胺的合成方法与流程
本发明属于化学技术领域,具体涉及一种阿奇霉素重排杂质内酰胺的合成方法。
背景技术:
阿奇霉素化学名称为-(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-2-乙基-3,4,10-三羟基-3,5,6,8,10,12,14-七甲基-11-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-1-氧杂-6-氮杂环十五烷-15酮,作为第二代大环内酯类抗生素,与红霉素相比,阿奇霉素的化学稳定性增强,降低了红霉素因为酸性降解而失去活性的问题,提高了血药浓度,大大延长了半衰期,被用于呼吸道、皮肤、泌尿系统和软组织感染的药物。该药已被列入《国家基本药品名录》,具有广阔的市场前景。目前我国年产阿奇霉素超过1000吨,是世界上阿奇霉素主要的生产国。
阿奇霉素内酰胺杂质是红霉素肟重排反应的副产物,含量极低,其未被收录于中国药典、美国药典及欧洲药典。经检索合成该杂质的方法主要有两种:一、乙醚作溶剂,红霉素肟在吡啶和对甲苯磺酰氯催化下重排反应得到内酰胺(djokic,slobodan;kobrehel,gabrijela;lazarevski,gorjana;lopotar,nevenka;tamburasev,zrinka;etal.;journalofthechemicalsociety,perkintransactions1:organicandbio-organicchemistry,1986;p.1881-1890),该方法运用了恶臭刺激性的吡啶和对甲苯磺酰氯,且收率只有63%;二、红霉素a6,9-亚胺醚分别在在质子性溶剂硝基甲烷和醋酸催化下分步反应约40小时生成内酰胺,分步操作复杂且收率只有55%(wilkening,robertr.;ratcliffe,ronaldw.;doss,georgea.;mosley,ralpht.;ball,richardg.;tetrahedron;vol.53;nb.50;1997;p.16923-16944)。因此开发一条高效安全合成该杂质的方法尤为重要。
技术实现要素:
为了解决上述技术问题,本发明提出了一种阿奇霉素重排杂质内酰胺的合成方法。
本发明技术方案:
一种阿奇霉素重排杂质内酰胺的合成方法,阿奇霉素重排杂质内酰胺结构如下:
所述阿奇霉素重排杂质内酰胺按照以下路线进行合成:
主要包含以下两个步骤:
3)化合物ⅱ加入溶剂1,试剂2,试剂3升温搅拌反应;加水洗涤,分层,取有机层,减压蒸馏回收有机层溶剂1,得杂质粗品;
4)精制:将杂质粗品中加入溶剂4重结晶得到阿奇霉素重排杂质内酰胺;
所述化合物ⅱ为(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮。
优选地,所述步骤1)溶剂1为二氯甲烷,试剂2为冰醋酸,试剂3为碳酸钾;
优选地,所述化合物ⅱ与二氯甲烷的固液比为1:4~6;
优选地,所述化合物ⅱ与冰醋酸的摩尔比为1:1.1~1.3。
优先地,所述化合物ⅱ与碳酸钾的摩尔比为1:1.1~1.3。
优选地,所述步骤1)反应温度为20~25℃,反应时间为2~3h。
优选地,所述步骤2)溶剂4为精制溶剂丙酮,杂质粗品与丙酮的固液比为1:0.4~0.6。
本发明有益效果如下:
1、提供了一种合成阿奇霉素重排杂质内酰胺-(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的方法,通过对其性质的研究,有利于对该杂质定性和定量控制,提高阿奇霉素原料药的质量。该方法操作简单,收率高,适合工业化生产。
2、以冰醋酸和碳酸钾作为重排试剂,其用量为1.1~1.3eq,反应效率高,反应后处理简单。
3、以二氯甲烷为溶剂,原料溶解度高,沸点低,容易蒸馏除去。
4、化合物ⅱ与二氯甲烷的固液比为1:4~6,降低醋酸浓度,防止原料酸降解反应的发生。
5、控制反应温度为20~25℃,加快重排反应速率,温度过高利于原料酸降解反应。
附图说明
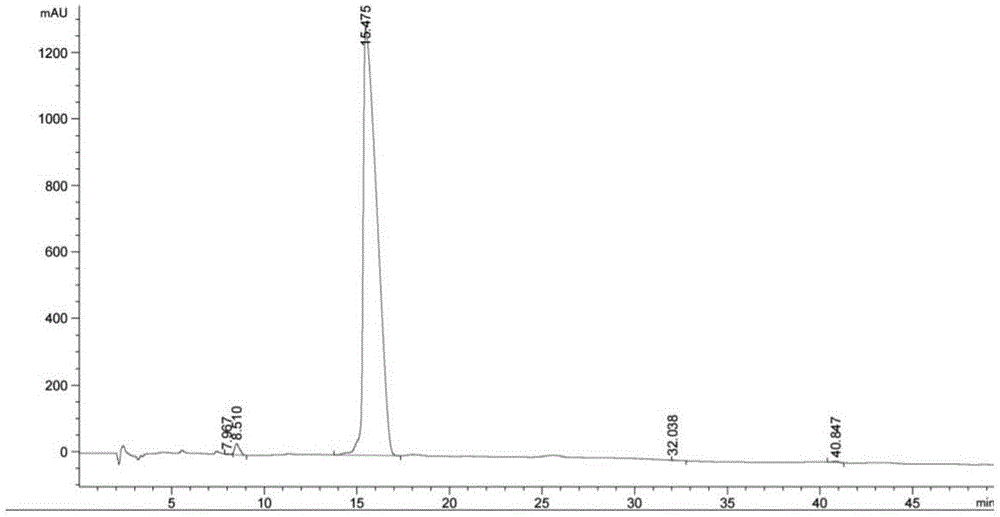
图1阿奇霉素重排杂质r的hplc图谱;
图2阿奇霉素重排杂质r的1h-nmr图谱;
图3阿奇霉素重排杂质r的13c-nmr图谱;
图4阿奇霉素重排杂质r的1h-1hcosy图谱;
图5阿奇霉素重排杂质r的hmbc图谱。
具体实施方式
实施例1(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷100ml,加入到三颈瓶中,控温20~25℃,加入冰醋酸1.72g(28.6mmol),碳酸钾3.95g(28.6mmol)溶于20ml水,搅拌反应2~3h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体18.6g,收率93%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮5ml,打浆1h,抽滤,烘干,得白色固体9.1g,收率91%,纯度99.5%。
实施例2(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷50ml,加入到三颈瓶中,控温20~25℃,加入冰醋酸1.72g(28.6mmol),碳酸钾3.95g(28.6mmol)溶于20ml水,搅拌反应2~3h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体17g,收率85%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮5ml,打浆1h,抽滤,烘干,得白色固体7.6g,收率76%,纯度98.2%。
实施例3(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷100ml,加入到三颈瓶中,控温40~45℃,加入冰醋酸1.72g(28.6mmol),碳酸钾3.95g(28.6mmol)溶于20ml水,搅拌反应2~3h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体16.2g,收率81%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮5ml,打浆1h,抽滤,烘干,得白色固体7.2g,收率72%,纯度98.1%。
实施例4(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷100ml,加入到三颈瓶中,控温20~25℃,加入冰醋酸3.44g(57.2mmol),碳酸钾7.9g(57.2mmol)溶于20ml水,搅拌反应2~3h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体17.8g,收率89%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮5ml,打浆1h,抽滤,烘干,得白色固体8.9g,收率89%,纯度99.2%。
实施例5(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷100ml,加入到三颈瓶中,控温20~25℃,加入冰醋酸1.72g(28.6mmol),碳酸钾3.95g(28.6mmol)溶于20ml水,搅拌反应1h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体17.6g,收率88%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮5ml,打浆1h,抽滤,烘干,得白色固体8.5g,收率85%,纯度98.7%。
实施例6(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的合成
将(3r,4r,5s,6r,9r,10s,11s,12r,13r,15r,z)-12-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-6-乙基-4,5-二羟基-10-[(2,6-二脱氧-3-c-甲基-3-o-甲基-a-l-核-已吡喃糖基)氧]-3,5,9,11,13,15-六甲基-7,16-二氧-2-氮杂双环[11.2.1]十六烷-1-烯-8-酮20g(化合物ⅱ,26mmol),二氯甲烷100ml,加入到三颈瓶中,控温20~25℃,加入冰醋酸1.72g(28.6mmol),碳酸钾3.95g(28.6mmol)溶于20ml水,搅拌反应2~3h,加入20ml水洗涤,分层,取有机层减压蒸馏得白色固体18.6g,收率93%。(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-11-[[(3,4,6)-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-2-乙基-3,4,10-三羟基-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-α-l-核-已吡喃糖基)氧]-3,5,8,10,12,14-六甲基-1-氧杂-6-氮杂环十五烷-7,15-二酮的精制
将上述粗品10g加入到单口瓶中,加入丙酮10ml,打浆1h,抽滤,烘干,得白色固体8.1g,收率81%,纯度99.6%。
综上所诉,实施案例1,为最优方案,粗品收率93%,干精品收率91%,纯度99.5%。
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!