氧化还原改性植物纤维的热塑性和韧性调控方法及其应用与流程
本发明属于天然高分子材料领域,特别涉及一种氧化还原改性植物纤维的热塑性和韧性调控方法及其应用。
背景技术:
20世纪以来,大量石油基高分子材料的生产造成了石油资源的短缺和严重的环境污染。以储量丰富、来源广泛、可生物降解的植物纤维原料制备具备优异性能的绿色高分子材料,从而部分替代石油基聚合物,将有效解决当前日益严重的资源和环境问题。
在长期的进化中,植物纤维的三种组分交织成复杂而致密的微纳复合结构。这种复杂机构使得其不能在高温下熔融加工,因为其在熔融之前,三大组分就已经发生了降解。传统的酯化、醚化虽然可实现植物纤维的塑化加工,但改性过程中大量酸酐和有机溶剂的使用不仅会污染环境、提高设备成本和回收成本,而且改性过程中会引起材料较大程度的降解,导致材料力学性能较差。
针对上述问题,cn201710662662.9木质纤维素塑化改性的方法及其应用,对植物纤维原料进行氧化还原反应,可较好地保留材料本身的强度,植物纤维经塑化改性后的玻璃化转变温度在90~110℃之间,从而实现植物纤维改性材料的热塑化加工。但是,该方法加工温度仍然较高,加工温度窗口依旧较窄,且制备得到的植物纤维材料的刚性较强,而韧性较差。
技术实现要素:
基于此,有必要提供一种氧化还原改性植物纤维的热塑加工性能和韧性的调控方法,使得处理后得到的植物纤维热塑加工性能得到进一步改善,可以在较低温度热压成型,在扩宽加工温度窗口的同时提高材料的韧性。
为实现上述目的,本发明提供了如下技术方案:
一种氧化还原改性植物纤维的热塑性和韧性调控方法,包括以下步骤:
(1)将植物纤维进行前处理;
(2)对前处理后的植物纤维进行氧化改性,对氧化改性的植物纤维进行还原改性,得氧化还原改性植物纤维;
(3)将增塑剂与氧化还原改性植物纤维混合均匀;所述增塑剂为羟基类增塑剂、离子液体类增塑剂、低共熔溶剂类增塑剂、酯类增塑剂、胺类增塑剂、缩水甘油醚类增塑剂或无机盐类增塑剂。
本发明还提供了一种具备热塑性和韧性的改性植物纤维材料,具体技术方案如下:
一种具备热塑性和韧性的改性植物纤维材料,由如上所述的方法处理后的植物纤维,通过干燥、热压制备得到。
基于上述技术方案,本发明具有以下有益效果:
本发明针对改性植物纤维原料加工温度较高,加工温度窗口较窄,材料韧性较差的问题,在氧化还原塑化改性的基础上,结合植物纤维这一特定基材的结构特性,合理地利用增塑剂,其中氧化还原改性纤维素和半纤维素开环以提高纤维素和半纤维素分子链的运动能力,再通过添加羟基类、离子液体类或低共熔溶剂类等增塑剂能进一步有效地破坏分子内和分子间的氢键作用,该增塑剂与植物纤维具备良好的相容性,在特定基材、氧化还原改性以及特定类型的增塑剂的配合下,扩展了植物纤维塑化加工温度窗口的同时,使其具备更好的加工流动性,还大幅度地提升材料的热塑性和韧性。
本发明制备得到的热塑性和韧性的氧化还原改性植物纤维材料还具备成本低廉、可生物降解、环境友好等特点,是一种生物质资源的高值化利用方式,具有广阔的研究前景。本发明大大拓展了全组分植物纤维材料的应用范围,对现有的部分石油基材料能起到一定的替代作用。
附图说明
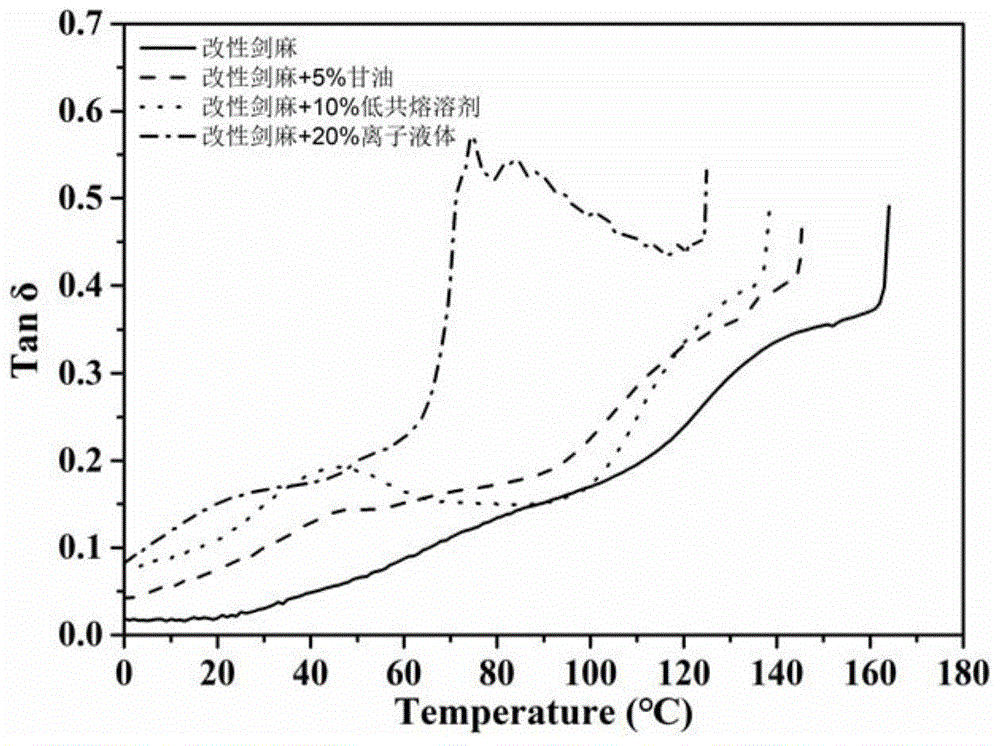
图1为实施例1、实施例2、实施例3和对比例1的损耗因子曲线图;
图2为实施例4、实施例5、实施例6和对比例2的损耗因子曲线图;
图3为实施例1制备的具备热塑性和韧性的改性剑麻纤维材料;
图4为对比例1制备的未添加增塑剂氧化还原改性剑麻纤维材料。
具体实施方式
为了便于理解本发明,下面将参照实施例对本发明进行更全面的描述,以下给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。应理解,下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用试剂,均为市售产品。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
本发明的一种氧化还原改性植物纤维的热塑性和韧性调控方法,包括以下步骤:(1)将植物纤维进行前处理;(2)对前处理后的植物纤维进行氧化改性,对氧化改性的植物纤维进行还原改性,得氧化还原改性植物纤维;(3)将增塑剂与氧化还原改性植物纤维混合均匀;所述增塑剂为羟基类增塑剂、离子液体类增塑剂、低共熔溶剂类增塑剂、酯类增塑剂、胺类增塑剂、缩水甘油醚类增塑剂或无机盐类增塑剂。
其中,本发明中所述增塑剂选自羟基类增塑剂、离子液体类增塑剂、低共熔溶剂类增塑剂、酯类增塑剂、胺类增塑剂、缩水甘油醚类增塑剂或无机盐类增塑剂。
其中,可选地,所述羟基类增塑剂包括:丙三醇、乙二醇、山梨醇或木糖醇。
可选地,所述离子液体类增塑剂包括:1-丁基-3-甲基咪唑氯盐、1-丁基-3-甲基咪唑乙酸盐、四氟硼酸n-乙基吡啶或四氟硼酸n-乙基吡啶。
可选地,所述低共熔溶剂类增塑剂为氢键受体和氢键供体的混合物;所述氢键受体包括:氯化胆碱或甜菜碱;所述氢键供体包括:尿素、硫脲、甘油、丁二醇、木糖醇或氨基酸、葡萄糖。
可选地,所述脂类增塑剂包括:邻苯二甲酸二乙酯、柠檬酸三甲酯、碳酸乙烯酯或碳酸丙烯酯。
可选地,所述胺类增塑剂包括:尿素、乙酰胺或氨基酸。
可选地,所述缩水甘油醚类增塑剂包括:缩水甘油醚、乙二醇一缩水甘油醚或丙三醇二缩水甘油醚。
可选地,所述无机盐类增塑剂包括:氯化锌、氯化锂或氯化铁。
优选地,所述增塑剂为:羟基类增塑剂、离子液体类增塑剂或低共熔溶剂类增塑剂。
其主要原理为:植物纤维的聚集态为纤维素、半纤维素和木质素相互交织而成的致密微纳结构,纤维素、半纤维素和木质素三大组分均为极性高分子,分子内和分子间存在着强烈氢键作用,且纤维素具备较高的结晶度。材料的特性影响增塑剂的选择,本发明采用的增塑剂在氧化还原改性纤维素和半纤维素开环以提高纤维素和半纤维素分子链的运动能力的基础上,进一步有效地破坏分子内和分子间的氢键作用,且增塑剂与植物纤维具备良好的相容性,在两者的配合下,使得植物纤维材料具备更好的加工流动性,在扩展塑化加工温度窗口的同时,还大幅度地提升材料的热塑性和韧性。
优选地,所述增塑剂的添加量与所述氧化还原改性植物纤维的重量比为(0.01~0.5):1。更优选地,所述增塑剂的添加量与所述氧化还原改性植物纤维的重量比为(0.05~0.25):1。更优选地,所述增塑剂为丙三醇(甘油)时,增塑剂与氧化还原改性植物纤维的重量比为(0.04~0.2):1,可进一步优选为(0.05~0.15):1;所述增塑剂为氯化胆碱和木糖醇组成的低共熔溶剂时,增塑剂与氧化还原改性植物纤维的重量比为(0.05~0.25):1,可进一步优选为(0.1~0.2):1;所述增塑剂为1-丁基-3-甲基咪唑氯盐时,增塑剂与氧化还原改性植物纤维的重量比为(0.05~0.25):1,可进一步优选为(0.1~0.2):1。
可选地,所述植物纤维选自:木本纤维、禾本科纤维、叶部纤维、树皮类纤维、麻类纤维、种毛纤维。合适的植物纤维与增塑剂的配合,降低了植物纤维的玻璃化转变温度,在使植物纤维材料具备更好的加工流动性、更宽的加工温度窗口的同时,还大幅度地提升材料的热塑性和韧性。
在其中一些实施方式中,采用高碘酸盐类氧化剂和硼氢化物类还原剂对植物纤维进行改性。优选地,氧化剂溶液为高碘酸盐的水溶液;和/或还原剂溶液为硼氢化物的水溶液。其中,所述高碘酸盐与所述植物纤维的重量比为(0.5~10):1;优选地,加入高碘酸盐的水溶液后,搅拌反应,反应的条件为:反应温度为20~100℃,反应时间为1~48h。更优选地,所述高碘酸盐的水溶液为高碘酸钠溶液。另外,所述硼氢化物与植物纤维的重量比为(0.1~1):1;优选地,加入硼氢化物的水溶液后,搅拌反应,反应的条件为:反应温度为20~40℃,反应时间为1~6h。更优选地,所述硼氢化物的水溶液为硼氢化钠溶液。结合合适的原料比例和反应条件,植物纤维原料在氧化还原反应的作用下,有利于纤维素和半纤维素的开环,降低纤维素和半纤维素分子链的刚性,同时减少分子内和分子间的氢键作用、降低纤维素的结晶度,有利于小分子增塑剂更好地嵌入植物纤维分子链间以提高热塑化效果。
优选地,步骤(1)所述的植物纤维的前处理还包括:将植物纤维破碎或切断后,进行蒸汽爆破预处理。更优选地,对植物纤维的前处理中,将植物纤维破碎至尺寸范围为5~40目或切断至长度为5~10mm,再进行蒸汽爆破预处理。从而更加有利于小分子增塑剂更好地嵌入植物纤维分子链间以提高热塑化效果。
本发明还提供了一种氧化还原改性植物纤维材料,由上述制备方法对植物纤维材料进行改性处理后,经过热压成型即可得到。其中,热压成型包括:加入增塑剂后的氧化还原改性剑麻纤维混合液在塑料盒中铺装好后进行冷冻干燥处理后,得到型胚,将型胚置入模具,先进行预压排气处理,然后进行热压成型,脱模即获得具备热塑性和韧性的改性剑麻纤维材料。所述热压成型中的工艺条件为:成型温度为60~220℃,成型压力为1~15mpa,成型时间为1~15min。本发明中通过氧化还原改性植物纤维的热塑性和韧性调控方法对植物纤维进行处理后,在较低温度条件下也能够热压成型,实现了扩宽加工温度窗口,在较大温度范围内均能热压成型制备得到韧性好、性能优的植物纤维材料。
下面结合具体实施例对本发明进行更全面的说明:
实施例1
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm。
(2)将高碘酸钠与步骤(1)所述的切断的剑麻纤维以2:1的重量比加入水溶液中,配置成高碘酸钠浓度为20g/l的悬浮液。然后在60℃下避光搅拌反应4h,过滤,洗涤,得到氧化改性剑麻纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性剑麻纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性剑麻纤维。
(4)将甘油与步骤(3)所述的氧化还原改性剑麻纤维以0.05:1的质量比混合,室温下搅拌均匀,得到加入增塑剂后的氧化还原改性剑麻纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性剑麻纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为155℃,成型压力为3mpa,成型时间为5min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性剑麻纤维材料。
实施例2
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的剑麻纤维以2:1的重量比加入水溶液中,配置成高碘酸钠浓度为20g/l的悬浮液。然后在60℃下避光搅拌反应4h,过滤,洗涤,得到氧化改性蒸汽爆破剑麻纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破剑麻纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破剑麻纤维。
(4)将氯化胆碱和木糖醇按2:1的质量比组成的低共熔溶剂与步骤(3)所述的氧化还原改性蒸汽爆破剑麻纤维以0.1:1的质量比混合,室温下搅拌均匀,得到加入增塑剂后的氧化还原改性蒸汽爆破剑麻纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性蒸汽爆破剑麻纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为150℃,成型压力为3mpa,成型时间为5min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性剑麻纤维材料。
实施例3
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的剑麻纤维以2:1的重量比加入水溶液中,配置成高碘酸钠浓度为20g/l的悬浮液。然后在60℃下避光搅拌反应4h,过滤,洗涤,得到氧化改性蒸汽爆破剑麻纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破剑麻纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破剑麻纤维。
(4)将1-丁基-3-甲基咪唑氯盐与步骤(3)所述的氧化还原改性蒸汽爆破剑麻纤维以0.2:1的质量比混合,室温下搅拌均,得到加入增塑剂后的氧化还原改性蒸汽爆破剑麻纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性蒸汽爆破剑麻纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为85℃,成型压力为3mpa,成型时间为5min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性剑麻纤维材料。
对比例1
一种植物纤维材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的剑麻纤维以2:1的重量比加入水溶液中,配置成高碘酸钠浓度为20g/l的悬浮液。然后在60℃下避光搅拌反应4h,过滤,洗涤,得到氧化改性蒸汽爆破剑麻纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破剑麻纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破剑麻纤维。
(4)将步骤(3)所述的氧化还原改性蒸汽爆破剑麻纤维在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(5)将步骤(4)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为175℃,成型压力为10mpa,成型时间为5min的工艺条件进行热压成型,脱模即获得未加增塑剂的改性剑麻纤维材料。
对比例2
一种植物纤维材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm,蒸汽爆破预处理。
(2)将1-丁基-3-甲基咪唑氯盐与步骤(1)所述的蒸汽爆破剑麻纤维以0.2:1的质量比混合,室温下搅拌均,得到加入增塑剂后的蒸汽爆破剑麻纤维混合液。
(3)将步骤(2)所述的加入增塑剂后的蒸汽爆破剑麻纤维在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(4)将步骤(3)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为200℃,成型压力为10mpa,成型时间为5min的工艺条件进行热压,脱模。材料无法成型加工,热压制备得到的材料缺陷较多、纤维界面结合差、易碎。
实施例4
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用纤维切断机将剑麻纤维切断至5~10mm,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的桉木纤维以2.67:1的重量比加入水溶液中,配置成高碘酸钠浓度为26.7g/l的悬浮液。然后在50℃下避光搅拌反应6h,过滤,洗涤,得到氧化改性蒸汽爆破桉木纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破桉木纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破桉木纤维。
(4)将甘油与步骤(3)所述的氧化还原改性蒸汽爆破桉木纤维以0.1:1的质量比混合,室温下搅拌均匀,得到加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为170℃,成型压力为5mpa,成型时间为6min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性桉木纤维材料。
实施例5
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用破碎机将桉木纤维破碎至5~40目,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的桉木纤维以2.67:1的重量比加入水溶液中,配置成高碘酸钠浓度为26.7g/l的悬浮液。然后在50℃下避光搅拌反应6h,过滤,洗涤,得到氧化改性蒸汽爆破桉木纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破桉木纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破桉木纤维。
(4)将氯化胆碱和木糖醇按2:1的质量比组成的低共熔溶剂与步骤(3)所述的氧化还原改性蒸汽爆破桉木纤维以0.2:1的质量比混合,室温下搅拌均匀,得到加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为145℃,成型压力为5mpa,成型时间为6min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性桉木纤维材料。
实施例6
一种氧化还原改性植物纤维的热塑性和韧性调控方法及材料的制备方法,具体步骤如下:
(1)利用破碎机将桉木纤维破碎至5~40目,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的桉木纤维以2.67:1的重量比加入水溶液中,配置成高碘酸钠浓度为26.7g/l的悬浮液。然后在50℃下避光搅拌反应6h,过滤,洗涤,得到氧化改性蒸汽爆破桉木纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破桉木纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破桉木纤维。
(4)将1-丁基-3-甲基咪唑氯盐与步骤(3)所述的氧化还原改性蒸汽爆破桉木纤维以0.2:1的质量比混合,室温下搅拌均匀,得到加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液。
(5)将步骤(4)所述的加入增塑剂后的氧化还原改性蒸汽爆破桉木纤维混合液在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(6)将步骤(5)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为155℃,成型压力为5mpa,成型时间为6min的工艺条件进行热压成型,脱模即获得具备热塑性和韧性的改性桉木纤维材料。
对比例3
一种植物纤维材料的制备方法,具体步骤如下:
(1)利用破碎机将桉木纤维破碎至5~40目,蒸汽爆破预处理。
(2)将高碘酸钠与步骤(1)所述的蒸汽爆破预处理后的桉木纤维以2.67:1的重量比加入水溶液中,配置成高碘酸钠浓度为26.7g/l的悬浮液。然后在50℃下避光搅拌反应6h,过滤,洗涤,得到氧化改性蒸汽爆破桉木纤维。
(3)将硼氢化钠与步骤(2)所述的氧化改性蒸汽爆破桉木纤维以0.5:1的重量比加入水溶液中,配置成硼氢化钠浓度为5g/l的悬浮液。室温下搅拌反应4h,过滤,洗涤,得氧化还原改性蒸汽爆破桉木纤维。
(4)将步骤(3)所述的氧化还原改性蒸汽爆破桉木纤维在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(5)将步骤(4)所述的型胚置入模具,先进行预压排气处理,然后以热压温度成型温度为195℃,成型压力为15mpa,成型时间为6min的工艺条件进行热压成型,脱模即获得未加增塑剂的改性桉木纤维材料。
对比例4
一种植物纤维材料的制备方法,具体步骤如下:
(1)利用破碎机将桉木纤维破碎至5~40目,蒸汽爆破预处理。
(2)将步骤1)所述的蒸汽爆破桉木纤维在塑料盒中铺装好后进行24h的冷冻干燥处理,得到型胚。
(3)将步骤(2)所述的型胚置入模具,先进行预压排气处理,然后以成型温度为210℃,成型压力为15mpa,成型时间为6min的工艺条件进行热压,脱模。材料无法成型加工,热压制备得到的材料缺陷较多、纤维界面结合差、易碎。
表1实施例与对比例制备的植物纤维材料参数对比
由表1结果可见,实施例1~6制备植物纤维材料的热压温度明显低于对比例1~4中的热压温度,其主要原因在于,实施例中采用了合适的植物纤维材料,进行氧化还原改性并配合了合适的增塑剂,使材料能在更低温度条件下热压成型。而对比例1和对比例3中,没有加入增塑剂,对比例2中没有进行氧化还原改性,对比例4既没有进行氧化还原改性,也没有加入增塑剂,没有实现如本发明中所述的结合对全植物纤维作为特定基材的氧化还原改性与增塑剂有效地破坏植物纤维分子内和分子间的氢键作用的效果,因此对比例需要更高的热压温度加工。而对比例2和对比例4在200℃和210℃条件下热压制备得到的材料仍然缺陷较多、纤维界面结合差、易碎,无法实现如本发明实施例中制备得到的韧性好的材料。
动态热机械测试
对以上所有改性纤维材料进行动态热机械测试,测试方法如下:采用dma242e型动态力学分析仪(德国耐驰仪器公司)进行测试与分析。测试采用拉伸模式,振动频率为1hz,目标振幅为10μm,比例因子为1.1。测试温度范围为0℃~240℃(或者直到样品断裂),升温速率为3℃/min。
测试结果:实施例1、实施例2、实施例3和对比例1制得的改性剑麻纤维材料的损耗因子-温度曲线如图1所示。实施例4、实施例5、实施例6和对比例2制得的改性桉木纤维材料的损耗因子-温度曲线如图2所示。
损耗因子-温度曲线中出现的肩峰和主峰分别对应材料的玻璃化转变温度1和玻璃化转变温度2。其中,玻璃化转变温度1为:二醇纤维素和二醇半纤维素的链段运动;玻璃化转变温度2为:晶区间二醇纤维素和二醇半纤维素分子链级别的受限运动。具体结果如表2所示
表2玻璃化转变温度1、玻璃化转变温度2和加工温度范围
由表2可见,与对比例1相比,实施例1~3的制备方法的玻璃化转变温度和柔性分子链运动温度更低,使得其加工温度范围更宽,而对比例1中的玻璃化转变温度和柔性分子链运动温度较高,其加工温度最低值较高,使得其加工温度范围很窄。说明实施例1~3中的氧化还原改性使得材料具有一定的热塑化加工性能,然而加工温度范围仍很窄。在氧化还原改性的基础上增加增塑剂后,材料的玻璃化转变温度和柔性分子链运动温度根据增塑剂的种类和配比有着不同程度的下降,热塑性能明显改善,加工温度范围也因此大幅拓宽。
按照实施例1所述的方法,制备得到的具备热塑性和韧性的改性剑麻纤维材料如图3所示。按照实施例1所述的方法,制备得到的未添加增塑剂氧化还原改性剑麻纤维材料如图4所示。对比例2和对比例4制备得到的材料缺陷较多、纤维界面结合差、易碎。可见,通过本发明所述的方法制备得到的材料的柔韧性好。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对以上实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!