生产RNA的方法与流程
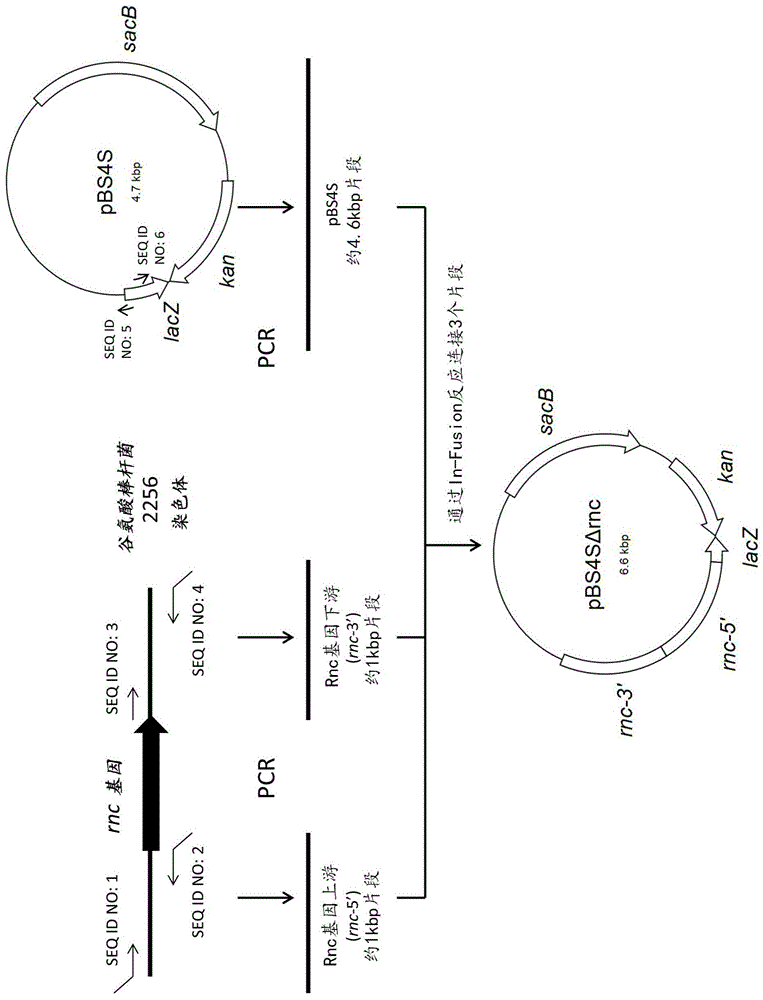
发明领域本发明涉及通过使用微生物发酵生产rna(核糖核酸)的方法。发明背景作为通过使用微生物生产rna的方法,已知例如使用缺乏核糖核酸酶iii(rna酶iii)的大肠杆菌(escherichiacoli)作为宿主在细菌细胞中积累rna的方法(非专利文献1)、与phi6噬菌体的功能组合使用丁香假单胞菌(pseudomonassyringae)作为宿主以在壳体中积累rna的方法(非专利文献2)、使用大肠杆菌作为宿主在细菌细胞中积累与trna的部分序列融合的rna的方法(非专利文献3)、使用大肠杆菌作为宿主在允许sirna与结合sirna的蛋白质形成复合物的情况下生产sirna的方法(非专利文献4)、使用小红卵菌属(rhodovulum)的细菌作为宿主以通过分泌产生来生产rna的方法(专利文献1)、以及使用酵母作为宿主在酵母线粒体中积累rna的方法(专利文献2)。引用表专利文献ptl1:专利文献1:wo2009/063969ptl2:专利文献2:wo2010/084371非专利文献npl1:非专利文献1:tenlladof,et.al.,crudeextractsofbacteriallyexpresseddsrnacanbeusedtoprotectplantsagainstvirusinfections.bmcbiotechnol.2003mar20;3:3.npl2:非专利文献2:aaltoap,et.al.,large-scaleproductionofdsrnaandsirnapoolsforrnainterferenceutilizingbacteriophagephi6rna-dependentrnapolymerase.rna.2007mar;13(3):422-9.npl3:非专利文献3:ponchonl,et.al.,agenericprotocolfortheexpressionandpurificationofrecombinantrnainescherichiacoliusingatrnascaffold.natprotoc.2009;4(6):947-59.npl4:非专利文献4:huangl,et.al.,efficientandspecificgeneknockdownbysmallinterferingrnasproducedinbacteria.natbiotechnol.2013apr;31(4):350-6.发明概述本发明的一个目的是提供一种有效生产rna的方法。本发明的发明人发现,通过使用缺乏核糖核酸酶iii(rna酶iii)的棒状杆菌细菌作为宿主,可以有效地生产rna,从而完成了本发明。因此,本发明可以例如如下实施。[1].一种生产目标rna的方法,所述方法包括:在培养基中培养具有所述目标rna的表达单元的棒状杆菌细菌(coryneformbacterium),以表达所述目标rna并在所述细菌的细胞中积累所述目标rna;并且从所述细胞中收集所述目标rna,其中所述细菌已经被修饰而使得与未经修饰的菌株相比核糖核酸酶iii的活性降低。[2].上述方法,其中所述核糖核酸酶iii是下述(a)、(b)或(c)中定义的蛋白质:(a)包含seqidno:52的氨基酸序列的蛋白质;(b)包含seqidno:52的氨基酸序列,且该氨基酸序列包含1至10个氨基酸残基的取代、缺失、插入和/或添加,并具有核糖核酸酶iii活性的蛋白质;(c)包含与seqidno:52的氨基酸序列显示90%或更高的同一性的氨基酸序列,并具有核糖核酸酶iii活性的蛋白质。[3].上述方法,其中通过减弱编码核糖核酸酶iii的基因的表达或通过破坏所述基因来降低核糖核酸酶iii的活性。[4].上述方法,其中通过缺失编码核糖核酸酶iii的基因来降低核糖核酸酶iii的活性。[5].上述方法,其中所述细菌具有拷贝数为5个拷贝/细胞或更多的所述表达单元。[6].上述方法,其中所述细菌具有拷贝数为70个拷贝/细胞或更多的所述表达单元。[7].上述方法,其中所述细菌具有含有所述表达单元的载体。[8].上述方法,其中所述表达单元在5’至3’的方向上含有在所述棒状杆菌细菌中起作用的启动子序列和编码所述目标rna的核苷酸序列。[9].上述方法,其中所述启动子序列是源自噬菌体的启动子。[10].上述方法,其中所述启动子序列是f1启动子或t7启动子。[11].上述方法,其中所述启动子序列是下述(a)或(b)中定义的启动子:(a)包含seqidno:13或78的核苷酸序列的启动子;(b)包含与seqidno:13或78的核苷酸序列显示90%或更高的同一性的核苷酸序列的启动子。[12].上述方法,其中所述细菌是属于棒杆菌属的细菌。[13].上述方法,其中所述细菌是谷氨酸棒杆菌(corynebacteriumglutamicum)。附图简述[图1]图显示了质粒ppbs4sδrnc的构建方案。[图2]用于确认rnc基因缺失的琼脂糖凝胶电泳图(照片)。[图3]图显示了质粒pvc7-sacb的构建方案。[图4]图显示了质粒pvc7-pf1-u1ainsert的构建方案。[图5]图显示了质粒的构建方案。(a)pvc7-pf1-hv-iap。(b)pvc7-pf1-hv-iap-pf1rev。[图6]琼脂糖凝胶电泳图(照片)显示了由于rnc基因缺失所致的rna生产的改善效果。[图7]琼脂糖凝胶电泳图(照片)显示了由于rna表达质粒的高拷贝数变异所致的rna生产的改善效果。[图8]图显示了质粒pl4440-pt7-u1ainsert的构建方案。[图9]图显示了质粒pl4440-pt7-hv-iap-pt7rev的构建方案。[图10]琼脂糖凝胶电泳图(照片)显示了在谷氨酸棒杆菌中对f1-启动子-表达系统观察到的rna生产量与大肠杆菌中对t7-启动子诱导表达系统观察到的rna生产量之间的比较结果。[图11]图显示了质粒ppk4xb-t7ter的构建方案。[图12]图显示了质粒ppk-t7lac的构建方案。[图13]图显示了质粒ppk-t7lac-vd-antiolac的构建方案。[图14]图显示了质粒pvc54的构建方案。[图15]图显示了质粒pbs5t-ptrb*的构建方案。[图16]图显示了质粒pbs5t-ptrb*-2ter的构建方案。[图17]图显示了质粒pbs5t-ptrb*-t7pol的构建方案。[图18]图显示了质粒pvc54-t7pol的构建方案。[图19]琼脂糖凝胶电泳图(照片)显示了在谷氨酸棒杆菌中通过使用t7-启动子诱导的表达系统生产rna。泳道1-2和6-7,克隆a;泳道3-4和8-9,克隆b;和泳道5,century-plusrna标志物(ambionam7145)。[图20]图显示了质粒ppk4-t7pol的构建方案。[图21]图显示了质粒pvc7-pt7-hv-iap-pt7rev的构建方案。[图22]琼脂糖凝胶电泳图(照片)显示了在谷氨酸棒杆菌中通过使用t7-启动子诱导的表达系统生产rna。[图23]琼脂糖凝胶电泳图(照片)显示了使用高拷贝数质粒ppk4h1生产rna。发明详述在下文中,将详细解释本发明。本发明的方法是生产目标rna的方法,该方法包括在培养基中培养具有目标rna的表达单元的棒状杆菌细菌,并收集转录的目标rna,其中细菌已经被修饰而使得核糖核酸酶iii的活性降低。用于此方法的棒状杆菌细菌也称为“本发明的细菌”。<1>本发明的细菌本发明的细菌是具有目标rna表达单元的棒状杆菌细菌,其已经被修饰而使得核糖核酸酶iii的活性降低。可以通过对棒状杆菌细菌应用目标rna的表达单元导入和核糖核酸酶iii的活性降低来获得本发明的细菌。在本发明中,用于构建本发明的细菌的修饰可以以任意顺序进行。也就是说,本发明的细菌可以通过例如将目标rna的表达单元导入棒状细菌,然后以核糖核酸酶iii的活性得以降低的方式修饰棒状细菌来获得。本发明的细菌也可以通过例如以核糖核酸酶iii的活性得以降低的方式修饰棒状杆菌,然后将目标rna的表达单元导入棒状杆菌来获得。本发明的细菌或构成本发明的细菌的细菌也称为“宿主”。本发明的细菌具有生产目标rna的能力(目标rna生产能力)。具体地,本发明的细菌至少由于细菌具有目标rna的表达单元而具有目标rna生产能力。例如,本发明的细菌可以是由于导入目标rna的表达单元,或者由于导入目标rna的表达单元和核糖核酸酶iii的活性降低的组合而获得目标rna生产能力的细菌。也就是说,要用于构建本发明的细菌并且在以核糖核酸酶iii的活性得以降低的方式进行修饰之前的菌株可以能够或不可以能够生产目标rna,假设该菌株具有目标rna的表达单元。只要细菌具有目标rna生产能力,本发明的细菌可以具有任何特征。例如,除了含有目标rna的表达单元的载体之外,本发明的细菌可以具有或不具有载体,例如质粒。也就是说,例如,当本发明的细菌固有具有质粒时,可以消除(除去)质粒。<1-1>具有目标rna生产能力的细菌在本发明中,术语“具有目标rna生产能力的细菌”指具有以下述程度表达和在细菌细胞中积累目标rna,使得当在培养基中培养细菌时可以收集目标rna的能力的细菌。具有目标rna生产能力的细菌可以是能够以比用未经修饰的菌株可获得的量更大的量在细菌细胞中积累目标rna的细菌。术语“未经修饰的菌株”指尚未以核糖核酸酶iii的活性得以降低的方式进行修饰的对照菌株。也就是说,未经修饰的菌株的实例包括野生型菌株和亲本菌株。具有目标rna生产能力的细菌也可以是能够在细菌细胞中以1mg/l培养物或更多、2mg/l培养物或更多、5mg/l培养物或更多、10mg/l培养物或更多、20mg/l培养物或更多、50mg/l培养物或更多、或100mg/l培养物或更多的量积累目标rna的细菌。在本发明中,棒状杆菌细菌用作宿主。棒状杆菌细菌的实例包括属于棒杆菌属、短杆菌属(brevibacterium)、分枝杆菌属(mycobacterium)、微杆菌属(microbacterium)等的细菌。棒状杆菌细菌的具体实例包括以下物种。嗜乙酰乙酸棒状杆菌(corynebacteriumacetoacidophilum)醋谷氨酸棒杆菌(corynebacteriumacetoglutamicum)解烷棒状杆菌(corynebacteriumalkanolyticum)帚石南棒状杆菌(corynebacteriumcallunae)钝齿棒状杆菌(corynebacteriumcrenatum)谷氨酸棒杆菌(corynebacteriumglutamicum)百合棒杆菌(corynebacteriumlilium)栖糖蜜棒杆菌(corynebacteriummelassecola)热产氨棒状杆菌(corynebacteriumthermoaminogenes)(有效棒杆菌(corynebacteriumefficiens))力士棒杆菌(corynebacteriumherculis)二歧短杆菌(brevibacteriumdivaricatum)(谷氨酸棒杆菌)黄色短杆菌(brevibacteriumflavum)(谷氨酸棒杆菌)未成熟短杆菌(brevibacteriumimmariophilum)乳糖发酵短杆菌(brevibacteriumlactofermentum)(谷氨酸棒杆菌)玫瑰色短杆菌(brevibacteriumroseum)解糖短杆菌(brevibacteriumsaccharolyticum)硫殖短杆菌(brevibacteriumthiogenitalis)产氨短杆菌(corynebacteriumammoniagenes)(corynebacteriumstationis)白色短杆菌(brevibacteriumalbum)蜡状短杆菌(brevibacteriumcerinum)嗜氨细杆菌(microbacteriumammoniaphilum)棒状杆菌细菌的具体实例可以包括以下菌株。嗜乙酰乙酸棒杆菌(corynebacteriumacetoacidophilum)atcc13870醋谷氨酸棒杆菌(corynebacteriumacetoglutamicum)atcc15806解烷棒杆菌(corynebacteriumalkanolyticum)atcc21511帚石南棒杆菌(corynebacteriumcallunae)atcc15991钝齿棒杆菌(corynebacteriumcrenatum)as1.542谷氨酸棒杆菌atcc13020,atcc13032,atcc13060,atcc13869,fermbp-734百合棒杆菌(corynebacteriumlilium)atcc15990栖糖蜜棒杆菌(corynebacteriummelassecola)atcc17965有效棒杆菌(corynebacteriumefficiens)(热产氨棒状杆菌(corynebacteriumthermoaminogenes))aj12340(fermbp-1539)力士棒杆菌(corynebacteriumherculis)atcc13868二歧短杆菌(brevibacteriumdivaricatum)(谷氨酸棒杆菌)atcc14020黄色短杆菌(brevibacteriumflavum)(谷氨酸棒杆菌)atcc13826,atcc14067,aj12418(fermbp-2205)未成熟短杆菌(brevibacteriumimmariophilum)atcc14068乳糖发酵短杆菌(brevibacteriumlactofermentum)(谷氨酸棒杆菌)atcc13869玫瑰色短杆菌(brevibacteriumroseum)atcc13825解糖短杆菌(brevibacteriumsaccharolyticum)atcc14066硫殖短杆菌(brevibacteriumthiogenitalis)atcc19240产氨短杆菌(corynebacteriumammoniagenes)(停滞棒杆菌(corynebacteriumstationis))atcc6871,atcc6872白色短杆菌(brevibacteriumalbum)atcc15111蜡状短杆菌(brevibacteriumcerinum)atcc15112嗜氨细杆菌(microbacteriumammoniaphilum)atcc15354棒状杆菌细菌包括先前被分类为短杆菌属但目前并入棒状杆菌属中的细菌(int.j.syst.bacteriol.,41,255(1991))。此外,停滞棒杆菌包括先前已被分类为产氨棒状杆菌,但目前基于16srrna的核苷酸序列分析等重新分类为停滞棒杆菌的细菌(int.j.syst.evol.microbiol.,60,874-879(2010))。这些菌株可从例如美国典型培养物保藏中心(地址:12301parklawndrive,rockville,maryland20852,p.o.box1549,manassas,va20108,unitedstatesofamerica)获得。也就是说,注册号被给予相应的菌株,并且菌株可以通过使用这些注册号订购(参见http://www.atcc.org/)。美国典型培养物保藏中心的目录中列出了菌株的注册号。这些菌株也可以从例如保藏菌株的保藏所获得。<1-2>导入目标rna的表达单元本发明的细菌具有目标rna的表达单元。通过将目标rna的表达单元导入棒状杆菌细菌,可以获得具有目标rna的表达单元的棒状杆菌细菌。术语“目标rna”指根据本发明的方法生产的rna。在本发明中,可以生产一种目标rna,或者可以产生两种或更多种目标rna。目标rna没有特别限制,只要它是宿主的外源rna,即只要它是除宿主的rna以外的rna即可。可以根据各种条件,例如目标rna的使用目的,适当选择目标rna。目标rna可以是例如天然存在的rna、其经修饰的rna或人工设计的rna。目标rna可以是例如源自微生物的rna、源自植物的rna、源自动物的rna或源自病毒的rna。目标rna可以是例如mrna(信使rna)、或非编码rna,例如rrna(核糖体rna)、trna(转移rna)、mirna(微小rna)、sirna(小干扰rna)、核酶和rna适体。例如,mrna可以是编码具有某种功能的蛋白质,例如酶、受体、转运蛋白、抗体、结构蛋白和调节因子的mrna,或编码不具有功能本身的蛋白质的mrna。顺便提及,本文提及的术语“蛋白质”包括所谓的肽,例如寡肽和多肽。例如,目标rna可以是具有如上所述的此类rna的任何核苷酸序列的rna。例如,目标rna也可以是具有如上所述的此类rna的任何核苷酸序列的部分序列的rna。例如,目标rna也可以是具有如上所述的此类rna的任何核苷酸序列的互补序列或其部分序列的rna。例如,目标rna也可以是具有如上所述的此类rna的任何核苷酸序列的变体序列、其部分序列或其互补序列的rna。关于后述核糖核酸酶iii基因的变体的描述可以在必要的变更后应用于此类变体序列。例如,目标rna也可以具有两种或更多种核苷酸序列的组合,所述核苷酸序列选自如上所述的此类rna的核苷酸序列、其部分序列、其互补序列及其变体序列。目标rna的具体实例包括茄二十八星瓢虫(henosepilachnavigintioctopunctata)的凋亡蛋白抑制剂的mrna的部分序列和构成科罗拉多马铃薯(coloradopotato)甲虫(马铃薯甲虫(leptinotarsadecemlineata))空胞中atp酶的亚单位a和e的mrna的部分序列。例如,目标rna可以是单链rna(由一个rna链分子组成的rna),或双链rna(由两个rna链分子组成的rna)。双链rna可以是由单一种类rna分子组成的双链(同双链),或由两种不同种类的rna分子组成的双链(异双链)。双链rna的具体实例包括例如由rna链及其互补链组成的双链rna。例如,目标rna也可以是由一个rna链分子和一个dna链分子组成的双链。目标rna可以包含单链区域和双链区域两者。也就是说,例如,单链rna可以在分子内部分地形成双链结构,例如茎-环结构。而且,例如,双链rna可以部分地含有单链结构。目标rna的长度没有特别限制。例如,目标rna的长度可以是10个残基或更多、20个残基或更多、50个或更多个残基、或100个残基或更多,或者可以是10000个残基或更少、5000个残基或更少、2000个残基或更少1000个残基或更少、或500个残基或更少,或可以是如定义的范围或其组合。术语“用于目标rna的表达单元”指配置成使得可以从其中表达目标rna的遗传构建体。目标rna的表达单元在5’至3’的方向上含有在棒状杆菌细菌中起作用的启动子序列和编码目标rna的核苷酸序列。启动子序列也简称为“启动子”。编码目标rna的核苷酸序列也称为“编码目标rna的基因”或“目标rna基因”。足够的是,将目标rna基因连接在启动子下游以使目标rna在启动子的控制下表达。目标rna的表达单元也可以在适当的位置处含有对于在棒状杆菌细菌中表达目标rna有效的调节序列(例如操纵基因和终止子),使得调节序列可以起作用。顺便提及,在本发明中,术语“目标rna基因的表达”、“目标rna基因的转录”、“目标rna的表达”和“目标rna的转录”可以彼此同义地使用。可以根据各种条件,例如目标rna的类型和转录模式,适当地设计目标rna的表达单元。目标rna的转录模式没有特别限制,只要获得目标rna。例如,目标rna基因可以在一个方向上转录(即通过使用双链的任意一条链作为模板),或在两个方向上转录(即通过使用双链的两条链作为模板)。目标rna基因在两个方向上的转录可以通过从相互相反方向插入基因排列的启动子(即在双链的各链中排列在基因5’侧的启动子)转录基因来进行。也就是说,目标rna的表达单元可以包含此类两个启动子。在此种情况下,两个启动子可以彼此相同或不同。通过在一个方向上转录目标rna基因,通常可以获得单链rna。通过在两个方向上转录目标rna基因,通常可以获得双链rna。双链rna也可以通过从其各自的表达单元转录双链rna的两条链来获得。目标rna基因可以通过例如克隆获得。对于克隆,例如,可以使用含有目标rna基因的核苷酸,例如基因组dna和cdna。目标rna基因也可以通过例如基于其核苷酸序列的全合成来获得(gene,60(1),115-127(1987))。得到的目标rna基因可以原样使用,或者根据需要进行修饰后使用。也就是说,可以通过修饰基因获得目标rna基因的变体。可以通过已知技术修饰基因。例如,可以通过位点特异性突变方法将目标突变引入dna的目标位点中。位点特异性突变方法的实例包括利用pcr的方法(higuchi,r.,61,于pcrtechnology,erlich,h.a.编,stocktonpress(1989);carter,p.,meth.于enzymol.,154,382(1987))和利用噬菌体的方法(kramer,w.andfrits,h.j.,meth.于enzymol.,154,350(1987);kunkel,t.a.etal.,meth.于enzymol.,154,367(1987))。或者,可以完全合成目标rna基因的变体。另外,目标rna的表达单元可以通过适当地对所获得的目标rna基因进行修饰,例如导入启动子序列来获得。顺便提及,可以以与目标rna基因相同的方式获得构成目标rna的表达单元的其他元件,例如启动子序列,或目标rna的整个表达单元。用于表达目标rna基因的启动子没有特别限制,只要其在宿主中起作用即可。术语“在宿主中起作用的启动子”指在宿主中显示启动子活性,即基因转录活性的启动子。启动子可以是源自宿主的启动子,或异源启动子。启动子可以是目标rna基因的天然启动子,或另一种基因的启动子。启动子可以是诱导型启动子或用于基因表达的组成性启动子。例如,启动子的实例包括糖酵解途径、磷酸戊糖途径、tca循环、氨基酸生物合成系统和细胞表面层蛋白的基因的启动子。作为启动子,也可以使用较强的启动子,例如下述的启动子。例如,较强启动子的实例包括人工修饰的p54-6启动子(appl.microbiol.biotechnol。,53,674-679(2000));可用乙酸、乙醇、丙酮酸等诱导的pta、acea、aceb、adh和amye启动子;和cspb、sod和tuf启动子,它们是有力的启动子(journalofbiotechnology,104(2003)311-323;appl.environ.microbiol.,2005dec;71(12):8587-96);以及lac启动子、tac启动子、trc启动子、f1启动子、t7启动子、t5启动子、t3启动子和sp6启动子。启动子的具体实例包括源自噬菌体的启动子,例如f1启动子、t7启动子、t5启动子、t3启动子和sp6启动子。启动子的更具体的实例包括f1启动子和t7启动子。f1启动子的核苷酸序列如seqidno:13所示。t7启动子的核苷酸序列如seqidno:78所示。作为启动子,也可以通过使用各种报告基因获得并使用高活性类型的现有启动子。例如,通过使启动子区域中的-35和-10区域更接近共有序列,可以增强启动子的活性(wo00/18935)。高活性类型启动子的实例包括各种tac样启动子(katashkinaji等,俄罗斯联邦专利申请号2006134574)和pnlp8启动子(wo2010/027045)。用于评估启动子强度的方法和强启动子的实例描述于goldstein等人的论文(prokaryoticpromotersinbiotechnology,biotechnol.annu.rev.,1,105-128(1995))等。启动子可以是具有上文例举的启动子的任何核苷酸序列(例如,如seqidno:13和78所示的核苷酸序列)的启动子。启动子也可以是上文例举的启动子的任何核苷酸序列(例如,如seqidno:13和78所示的核苷酸序列)的保守变体。也就是说,上文列举的启动子各自可以原样使用,或者在根据需要进行修饰后使用。用上述启动子名称定义的启动子不仅分别包括上文例举的启动子,而且还包括其保守变体。也就是说,例如,术语“f1启动子”不仅包括具有seqidno:13所示的核苷酸序列的启动子,而且还包括其保守变体。而且,例如,术语“t7启动子”不仅包括具有seqidno:78所示的核苷酸序列的启动子,而且还包括其保守变体。关于后面提到的核糖核酸酶iii基因的保守变体的描述可以在必要的变更后应用于启动子的保守变体。例如,启动子可以是具有核苷酸序列的启动子,所述核苷酸序列与seqidno:13或78所示的核苷酸序列显示例如80%或更多、优选90%或更多、更优选95%或更多、更优选97%或更多、特别优选99%或更多的同源性,只要维持原始功能。顺便提及,用于启动子的术语“原始功能”指在某些条件下提供紧接其下游连接的基因的表达的功能。术语“某些条件”指原始启动子提供紧接其下游连接的基因表达的条件。例如,可以从f1启动子中组成性表达基因。而且,例如,可以在t7rna聚合酶、t5rna聚合酶、t3rna聚合酶或sp6rna聚合酶的存在下从t7启动子、t5启动子、t3启动子或sp6启动子诱导表达基因。启动子的保守变体可以具有例如原始启动子的转录活性的80%或更多、90%或更多或100%或更多的转录活性。可以通过例如使用报告基因来确认基因表达的存在或缺乏以及基因表达的强度(转录活性)。终止基因转录的终止子可以位于目标rna基因的下游。终止子没有特别限制,只要它在宿主中起作用即可。终止子可以是源自宿主的终止子,或异源终止子。终止子可以是目标rna基因的天然终止子,或另一种基因的终止子。例如,终止子的具体实例包括噬菌体bfk20的终止子。在各种微生物中可获得的载体、启动子和终止子详细公开于“fundamentalmicrobiologyvol.8,geneticengineering,kyoritsushuppanco.,ltd,1987”中,并且可以使用那些。将目标rna的表达单元导入棒状杆菌细菌中的方法没有特别限制。术语“导入目标rna的表达单元”指使宿主含有目标rna的表达单元,并且可以具体指将目标rna基因表达性导入宿主中。术语“导入目标rna的表达单元”不仅包括共同导入已经预先构建到宿主中的目标rna的表达单元的情况,而且还包括至少将目标rna基因导入宿主中以在宿主中构建目标rna的表达单元的情况,除非另有说明。在本发明的细菌中,目标rna的表达单元可以存在于与染色体分开可自主复制的载体,例如质粒中,或者可以整合到染色体中。也就是说,例如,本发明的细菌可以在载体上具有目标rna的表达单元,换句话说,可以具有含有目标rna的表达单元的载体。例如,本发明的细菌也可以具有染色体上目标rna的表达单元。本发明的细菌可以仅具有目标rna的表达单元的一个拷贝,或目标rna的表达单元的两个或更多个拷贝。例如,本发明的细菌所具有的目标rna的表达单元的拷贝数可以是5个拷贝/细胞或更多、10个拷贝/细胞或更多、20个拷贝/细胞或更多、30个拷贝/细胞或更多、50个拷贝/细胞或更多、70个拷贝/细胞或更多、100个拷贝/细胞或更多、150个拷贝/细胞或更多、200个拷贝/细胞或更多、300个拷贝/细胞或更多、500个拷贝/细胞或更多、1000个拷贝/细胞或更多,或可以是2000个拷贝/细胞或更少、1500个拷贝/细胞或更少、1000个拷贝/细胞或更少、500个拷贝/细胞或更少、或300个拷贝/细胞或更少,或可以是定义为其不矛盾组合的范围。本发明的细菌可以仅具有一种目标rna的表达单元,或者具有两种或更多种目标rna的表达单元。目标rna的表达单元的拷贝数和种类也可以分别解读为目标rna基因的拷贝数和种类。当本发明的细菌具有两个或更多个目标rna的表达单元时,足够的是本发明的细菌含有那些表达单元,从而生产目标rna。例如,可以在单一表达载体上或染色体上含有所有那些表达单元。或者,可以分别在多个表达载体上,或者分别在单一或多个表达载体和染色体上含有那些表达单元。可以通过例如使用含有目标rna的表达单元的载体将目标rna的表达单元导入宿主中。含有目标rna的表达单元的载体也称为“目标rna的表达载体”。可以通过例如连接目标rna的表达单元与载体来构建目标rna的表达载体。或者,例如,当载体含有在棒状杆菌细菌中起作用的启动子时,也可以通过在启动子下游连接目标rna基因来构建目标rna的表达载体。通过用目标rna的表达载体转化宿主,可以获得已经导入载体的转化体,即,可以将目标rna的表达单元导入宿主中。作为载体,可以使用在宿主细胞中可自主复制的载体。载体优选是多拷贝载体。例如,载体的拷贝数可以是5拷贝/细胞或更多、10拷贝/细胞或更多、20拷贝/细胞或更多、30拷贝/细胞或更多、50拷贝/细胞或更多、70拷贝/细胞或更多、100拷贝/细胞或更多、150拷贝/细胞或更多、200拷贝/细胞或更多、300拷贝/细胞或更多、500拷贝/细胞或更多、1000拷贝/细胞或更多,或可以是2000拷贝/细胞或更少、1500拷贝/细胞或更少、1000拷贝/细胞或更少、500拷贝/细胞或更少、或300拷贝/细胞或更少,或可以是定义为其不矛盾组合的范围。此外,载体优选含有标志物,例如抗生素抗性基因或用于选择转化体的营养缺陷型互补基因。此外,载体可含有用于表达导入基因的启动子和/或终止子。例如,载体可以是源自细菌质粒的载体、源自酵母质粒的载体、源自噬菌体的载体、粘粒、噬菌粒等。可在棒状杆菌细菌中自主复制的载体的具体实例包括,例如,phm1519(agric.biol.chem.,48,2901-2903(1984));pam330(agric.biol.chem.,48,2901-2903(1984));通过对这些进行改进并且具有药物抗性基因获得的质粒;pcry30(日本专利公开文本(kokai)no.3-210184);pcry21、pcry2ke、pcry2kx、pcry31、pcry3ke和pcry3kx(日本专利公开文本(kokai)no.2-72876和美国专利no.5,185,262);pcry2和pcry3(日本专利公开文本(kokai)no.1-191686);paj655、paj611和paj1844(日本专利公开文本(kokai)no.58-192900);pcg1(日本专利公开文本(kokai)no.57-134500);pcg2(日本专利公开文本(kokai)no.58-35197);pcg4和pcg11(日本专利公开文本(kokai)no.57-183799);ppk4(美国专利no.6,090,597);pvk4(日本专利公开文本(kokai)no.9-322774);pvk7(日本专利公开文本(kokai)no.10-215883);pvk9(us2006-0141588);pvc7(日本专利公开文本(kokai)no.9-070291);和pvs7(wo2013/069634)。在棒状杆菌细菌中可自主复制的载体的具体实例还包括例如ppk4h1、ppk4h2、ppk4h3、ppk4h4、ppk4h5和ppk4h6(本实施例),它们是pvc7的变体。在棒状杆菌细菌中可自主复制的载体的具体实例还包括例如ppk4h1、ppk4h2、ppk4h3、ppk4h4、ppk4h5和ppk4h6(本实施例),它们是ppk4的变体。此外,可以通过例如使用转座子如人工转座子将目标rna的表达单元导入宿主的染色体中。当使用转座子时,可以通过同源重组或由于其转座活性将目标rna的表达单元导入染色体。也可以通过利用同源重组的导入方法将目标rna的表达单元导入宿主的染色体中。例如,利用同源重组的导入方法的实例包括使用线性dna、含有温度敏感性复制起点的质粒、能够接合转移的质粒、不具有在宿主中起作用的复制起点的自杀载体等的方法。可以仅导入目标rna的表达单元的一个拷贝或两个或更多个拷贝。例如,通过使用在染色体上以多个拷贝存在的序列作为靶物进行同源重组,可以将目标rna的表达单元的多个拷贝导入染色体中。在染色体上以多个拷贝存在的序列的实例包括重复dna和位于转座子的两个末端的反向重复序列。另外,可以至少将目标rna基因导入染色体中,以构建染色体上的目标rna的表达单元。例如,通过在宿主染色体上的启动子序列下游导入目标rna基因,可以在染色体上构建目标rna的表达单元。顺便提及,将目标rna的表达单元(例如目标rna基因)的一部分导入染色体中可以以与将目标rna的整个表达单元导入染色体相同的方式进行。转化方法没有特别限制,并且可以使用通常使用的方法,例如原生质体方法(gene,39,281-286(1985))、电穿孔方法(bio/technology,7,1067-1070(1989))和电脉冲方法(jph2-207791a)。<1-2>核糖核酸酶iii活性的降低本发明的细菌已经被修饰而使得核糖核酸酶iii(rnaseiii)的活性降低。具体地,本发明的细菌已经以与未经修饰的菌株相比核糖核酸酶iii的活性得以降低的方式进行了修饰。核糖核酸酶iii的活性可以降低至例如未修饰菌株的活性的50%或更低、20%或更低、10%或更低、5%或更低或0%。也就是说,本发明的细菌可以已经以例如核糖核酸酶iii的活性得以缺失(消除)的方式进行了修饰。通过以核糖核酸酶iii的活性得以降低的方式修饰棒状杆菌细菌,可以改善细菌的目标rna生产能力,也就是说,可以增加通过使用细菌生产目标rna。在下文中,将解释核糖核酸酶iii和编码它的基因。术语“核糖核酸酶iii”指具有催化切割特定rna(例如双链rna)的反应的活性的蛋白质。编码核糖核酸酶iii的基因也称为“核糖核酸酶iii基因”。核糖核酸酶iii基因的实例包括rnc基因。由rnc基因编码的蛋白质(核糖核酸酶iii)也称为“rnc蛋白质”。由棒状杆菌细菌所拥有的核糖核酸酶iii基因,例如rnc基因的核苷酸序列,以及由这些基因编码的核糖核酸酶iii,例如rnc蛋白的氨基酸序列可以从例如公共数据库如ncbi(国立生物技术信息中心)获得。谷氨酸棒杆菌atcc13869菌株的rnc基因的核苷酸序列和由该基因编码的rnc蛋白的氨基酸序列分别显示在seqidno:51和52中。也就是说,例如,核糖核酸酶iii基因可以是具有上文例举的任何核糖核酸酶iii基因的核苷酸序列(例如,seqidno:51所示的核苷酸序列)的基因。例如,核糖核酸酶iii也可以是具有上文例举的任何核糖核酸酶iii的氨基酸序列(例如,seqidno:52所示的氨基酸序列)的蛋白质。除非另有说明,表述“基因或蛋白质具有核苷酸或氨基酸序列”指基因或蛋白质包含所述核苷酸或氨基酸序列,并且也包括基因或蛋白质由所述核苷酸或氨基酸序列组成的情况。核糖核酸酶iii基因可以是上文例举的任何核糖核酸酶iii基因(例如,具有seqidno:51所示核苷酸序列的基因)的变体,只要维持原始功能即可。类似地,核糖核酸酶iii可以是上文例举的任何核糖核酸酶iii(例如,具有seqidno:52所示氨基酸序列的蛋白质)的变体,只要维持原始功能即可。维持原始功能的此类变体也称为“保守变体”。在本发明中,术语“rnc基因”不仅包括上文例举的rnc基因,而且还包括其保守变体。类似地,术语“rnc蛋白”不仅包括上文例举的rnc蛋白,而且还包括其保守变体。例如,保守变体的实例包括上文例举的核糖核酸酶iii基因和核糖核酸酶iii的同源物和人工修饰形式。表述“维持原始功能”指基因或蛋白质的变体具有与原始基因或蛋白质的功能(例如活性或性质)对应的功能(例如活性或性质)。也就是说,用于核糖核酸酶iii基因的表述“维持原始功能”指基因的变体编码维持原始功能的蛋白质(即具有核糖核酸酶iii活性的蛋白质)。此外,用于核糖核酸酶iii的表述“维持原始功能”指蛋白质的变体具有核糖核酸酶iii活性。核糖核酸酶iii活性可以通过例如将酶与充当其底物的rna(例如双链rna)一起温育,并测量rna的酶依赖性切割来测量。具体地,核糖核酸酶iii活性通常以下述方式测量(methodsenzymol.2001;342:143-58.)。一个实例是下述方法:将酶(例如来自细胞的粗提取物或其部分纯化的酶)添加到双链形式的经3h标记的聚(a-u)的合成底物,以在35℃对它们进行反应,用三氯乙酸处理反应混合物,并测量含有高分子量核苷酸的沉淀级份中反应时间依赖性的放射性降低程度。也就是说,核糖核酸酶iii活性可以基于放射性降低的程度作为底物切割的指标来计算。此外,另一个实例是下述方法:将32p-放射性标记的双链rna作为底物添加到含有酶的反应混合物(30mmtris-hcl(ph8.0)、250mm谷氨酸钾或160mmnacl、5mm亚精胺、0.1mmedta和0.1mmdtt),在37℃温育5分钟,对其添加终浓度10mm的mgcl2以引发rna切割反应,并在适当的反应进行后对其添加等体积的edta和电泳标志物染料的混合物以停止反应,其中edta浓度为提供20mm或更高终浓度的浓度。然后,可以通过将反应后的样品应用于用含有7m尿素的tbe缓冲液(89mmtris/tris-硼酸盐和2mmedta)使用变性15%(w/v)聚丙烯酰胺凝胶的电泳,并将凝胶应用于辐射成像分析仪以分析经切割的rna片段来检测核糖核酸酶iii活性。在下文中,将解释保守变体的实例。可以使用上文例举的任何核糖核酸酶iii基因的核苷酸序列或上文例示的任何核糖核酸酶iii的氨基酸序列作为查询序列通过例如blast搜索或fasta搜索从公共数据库容易地获得核糖核酸酶iii基因的同源物或核糖核酸酶iii的同源物。此外,可以通过例如使用棒状杆菌细菌的染色体作为模板和基于这些已知的核糖核酸酶iii基因的任一种及其相邻区域的核苷酸序列制备的寡核苷酸作为引物的pcr来获得核糖核酸酶iii基因的同源物。核糖核酸酶iii基因可以是编码具有上文例举的任何核糖核酸酶iii的氨基酸序列(例如,seqidno:52所示的氨基酸序列),且该氨基酸序列包括在一个或几个位置处一个或几个氨基酸残基取代、缺失,插入和/或添加的蛋白质的基因,只要维持原始功能即可。尽管上文提到的术语“一个或几个”所指的数字可以根据蛋白质的三维结构中的氨基酸残基的位置或氨基酸残基的类型而不同,具体地,例如它是1至50、1至40、或1至30,优选1至20,更优选1至10,还更优选1至5,特别优选1至3。一个或几个氨基酸残基的上述取代、缺失、插入和/或添加是维持蛋白质的正常功能的保守突变。保守突变的典型实例是保守取代。保守取代是下述突变,其中若取代位点是芳香族氨基酸,则取代在phe、trp和tyr之间相互发生;若它是疏水性氨基酸,则在leu、ile和val之间相互发生;若它是极性氨基酸,则在gln和asn之间相互发生;若它是碱性氨基酸,则在lys、arg和his之间相互发生;若它是酸性氨基酸,则在asp和glu之间相互发生;若它是具有羟基的氨基酸,则在ser和thr之间相互发生。具体地,认为是保守取代的取代的实例包括用ser或thr取代ala,用gln,his或lys取代arg,用glu,gln,lys,his或asp取代asn,用asn,glu或gln取代asp,用ser或ala取代cys,用asn,glu,lys,his,asp或arg取代gln,用gly,asn,gln,lys或asp取代glu,用pro取代gly,用asn,lys,gln,arg或tyr取代his,用leu,met,val或phe取代ile,用ile,met,val或phe取代leu,用asn,glu,gln,his或arg取代lys,用ile,leu,val或phe取代met,用trp,tyr,met,ile或leu取代phe,用thr或ala取代ser,用ser或ala取代thr,用phe或tyr取代trp,用his,phe或trp取代tyr,和用met,ile或leu取代val。此外,如上所述的氨基酸残基的此类取代、缺失、插入或添加包括由于个体差异,或该基因来源的细菌的物种差异(突变体或变体)而导致的天然存在的突变。核糖核酸酶iii基因可以是编码具有与上文例举的任何核糖核酸酶iii的总氨基酸序列显示例如80%或更多,优选90%或更多,更优选95%或更多,更优选97%或更多,特别优选99%或更多的同源性的氨基酸序列的蛋白质的基因,只要维持原始功能即可。在本说明书中,“同源性”意指“同一性”。核糖核酸酶iii基因也可以是能够在严格条件下与上文例举的任何核糖核酸酶iii基因的核苷酸序列(例如seqidno:51所示的核苷酸序列)的互补序列或可以从互补序列中制备的探针杂交的dna,只要维持原始功能即可。术语“严格条件”指形成所谓的特异性杂交体并且不形成非特异性杂交体的条件。严格条件的实例包括高度同源dna彼此杂交,例如,不小于80%同源,优选不小于90%同源,更优选不小于95%同源,更优选不小于97%同源,特别优选不小于99%同源的dna彼此杂交,且小于上述项的同源性的dna不彼此杂交的条件,或典型southern杂交的清洗条件,即在对应于1xssc,0.1%sds于60℃,优选0.1xssc,0.1%sds于60℃,更优选0.1xssc,0.1%sds于68℃的盐浓度和温度清洗一次,优选2或3次的条件。例如,探针可以是基因的互补序列的一部分。此类探针可以使用基于已知基因序列制备的寡核苷酸作为引物和含有这些核苷酸序列中任一种的dna片段作为模板通过pcr制备。作为探针,例如,可以使用长度为约300bp的dna片段。在此类情况下,杂交的清洗条件可以是例如50℃,2xssc和0.1%sds。此外,由于密码子的简并性随宿主而不同,核糖核酸酶iii基因中的任意密码子可以用相应的等同密码子替换。也就是说,核糖核酸酶iii基因可以是由于遗传密码的简并性所致的上文例举的任何核糖核酸酶iii基因的变体。两个序列之间的序列同一性的百分比可以通过例如使用数学算法来确定。此类数学算法的非限制性实例包括myersandmiller(1988)cabios4:11-17,thelocalhomologyalgorithmofsmithetal(1981)adv.appl.math.2:482的算法、needlemanandwunsch(1970)j.mol.biol.48:443-453的同源性比对算法、pearsonandlipman(1988)proc.natl.acad.sci.85:2444-2448和搜索同源性的方法和karlinandaltschul(1990)proc.natl.acad.sci.usa87:2264的算法的修改形式,诸如karlinandaltschul(1993)proc.natl.acad.sci.usa90:5873-5877中描述的。通过使用基于此类数学算法的程序,可以进行用于测定序列同一性的序列比较(即比对)。程序可以由计算机适当进行。此类程序的实例包括但不限于pc/gene程序的clustal(可获自intelligenetics,mountainview,calif.)、align程序(version2.0)和wisconsingeneticssoftwarepackage,version8(可获自geneticscomputergroup(gcg),575sciencedrive,madison,wis.,usa)的gap、bestfit、blast、fasta和tfasta。可以通过使用例如初始参数来进行使用这些程序的比对。clustal程序完全记载于higginsetal.(1988)gene73:237-244(1988),higginsetal.(1989)cabios5:151-153,corpetetal.(1988)nucleicacidsres.16:10881-90,huangetal.(1992)cabios8:155-65和pearsonetal.(1994)meth.mol.biol.24:307-331。为了获得与靶核苷酸序列同源的核苷酸序列,特别地,例如,可以通过使用得分为100和字长为12的blastn程序进行blast核苷酸搜索。为了获得与靶蛋白同源的氨基酸序列,特别地,例如可以通过使用得分为50和字长为3的blastx程序进行blast蛋白质搜索。对于blast核苷酸搜索和blast蛋白质搜索,参见http://www.ncbi.nlm.nih.gov。另外,为了比较的目的,可以使用gappedblast(blast2.0)以获得包括缺口的比对。另外,可以使用psi-blast进行重复搜索以检测序列之间的远距离关系。对于gappedblast和psi-blast,参见altschuletal.(1997)nucleicacidsres.25:3389。当使用blast、gappedblast或psi-blast时,可以使用每个程序的初始参数(例如对于核苷酸序列的blastn和对于氨基酸序列的blastx)。也可以手动进行比对。将两个序列之间的序列同一性计算为当比对两个序列以彼此最大限度匹配时两个序列中匹配的残基的比率。关于基因和蛋白质的变体的上述描述可以在必要的变更后应用于其他任意蛋白质和目标rna的变体,以及编码它们的基因。在下文中,将解释用于降低蛋白质(酶)如核糖核酸酶iii的活性的方法。表述“蛋白质的活性得以降低”指与未经修饰的菌株相比,蛋白质的活性得以降低。具体地,表述“蛋白质的活性得以降低”指与未经修饰的菌株相比,每个细胞的蛋白质活性得以降低。本文使用的术语“未经修饰的菌株”指尚未以目标蛋白质的活性得到降低的方式进行修饰的对照菌株。未经修饰的菌株的实例包括野生型菌株和亲本菌株。未经修饰的菌株的具体实例包括细菌物种的各种模式株。未经修饰的菌株的具体实例也包括上文关于棒状杆菌细菌的描述所例举的菌株。也就是说,在一个实施方案中,与模式株,即本发明的细菌所属的物种的模式株相比,蛋白质的活性可以得以降低。在另一个实施方案中,与谷氨酸棒杆菌atcc13032菌株相比,蛋白质的活性也可以得以降低。在另一个实施方案中,与谷氨酸棒杆菌2256菌株(atcc13869)相比,蛋白质的活性也可以得以降低。“蛋白质活性得以降低”的状态也包括蛋白质活性完全消失的状态。更具体地,表述“蛋白质的活性得以降低”可以指与未经修饰的菌株相比,每个细胞的蛋白质分子数目得以减少,和/或蛋白质的每个分子的功能得以降低。也就是说,表述“蛋白质活性得以降低”中的术语“活性”不限于蛋白质的催化活性,也可以指编码蛋白质的基因转录量(即mrna的量)或蛋白质的翻译量(即蛋白质的量)。“每个细胞的蛋白质分子数目得以减少”的状态也包括蛋白质根本不存在的状态。“蛋白质的每个分子的功能得以降低”的状态也包括每个蛋白质分子的功能已完全消失的状态。蛋白质活性降低的程度没有特别限制,只要与未经修饰的菌株相比活性得以降低即可。蛋白质的活性可以降低至例如未修饰菌株的50%或更小、20%或更小、10%或更小、5%或更小或0%。用于降低蛋白质活性的修饰可以通过例如降低编码蛋白质的基因的表达来实现。表述“基因的表达得以降低”指与未经修饰的菌株如野生型菌株和亲本菌株相比,基因的表达得以降低。具体地,表述“基因的表达得以降低”指与未经修饰的菌株相比,每个细胞的基因表达得以降低。更具体地,表述“基因的表达得以降低”可以指基因的转录量(即mrna的量)得以减少,和/或基因的翻译量(即从基因表达的蛋白质量)得以减少。“基因表达得以降低”的状态也包括基因完全不表达的状态。“基因表达得以降低”的状态也称为“基因表达得以减弱”。基因的表达可以降低至例如未修饰菌株的50%或更小、20%或更小、10%或更小、5%或更小或0%。基因表达的降低可以是由于例如转录效率的降低、翻译效率的降低或它们的组合。可以通过修饰基因的表达控制序列,例如启动子、shine-dalgarno(sd)序列(也称为核糖体结合位点(rbs))和rbs和基因的起始密码子之间的间隔区来降低降低基因的表达。当修饰表达控制序列时,优选修饰表达控制序列的一个或多个核苷酸,更优选两个或更多个核苷酸,特别优选三个或更多个核苷酸。例如,可以通过例如用较弱的启动子替换染色体上基因的启动子来降低基因的转录效率。术语“较弱的启动子”指与基因的固有存在的野生型启动子相比,提供基因的减弱转录的启动子。较弱启动子的实例包括例如诱导型启动子。也就是说,诱导型启动子可以在非诱导条件下,例如在不存在相应的诱导物的情况下作为较弱的启动子起作用。此外,可以缺失表达控制序列的一部分或全部。也可以通过例如操纵负责表达控制的因子来降低基因的表达。负责表达控制的因子的实例包括负责转录或翻译控制的低分子(诱导物,抑制剂等)、负责转录或翻译控制的蛋白质(转录因子等)、负责转录或翻译控制的核酸(sirna)等),等等。此外,也可以通过例如将降低基因表达的突变引入基因编码区中来降低基因的表达。例如,可以通过用在宿主中较少使用的同义密码子替换基因编码区中的密码子来降低基因的表达。此外,例如,如后所述,基因表达可以由于基因的破坏而得以降低。用于降低蛋白质活性的修饰也可以通过例如破坏编码蛋白质的基因来实现。表述“基因得以破坏”指基因以不产生能够正常起作用的蛋白质的方式进行修饰。“不产生正常起作用的蛋白质”的状态包括根本不从基因产生蛋白质的状态,以及从基因产生每个分子的功能(例如活性或性质)得以降低或消除的蛋白质的状态。可以通过例如缺失染色体上的基因来实现基因的破坏。术语“基因的缺失”指缺失基因编码区的部分或整个区域。此外,可以缺失包括染色体上基因编码区上游和下游序列的整个基因。待缺失的区域可以是任何区域,例如n端区域(编码蛋白质的n端区域的区域)、内部区域或c端区域(编码蛋白质的c端区域的区域),只要蛋白质的活性可以得以降低。较长区域的缺失通常可以更加确定地使基因失活。例如,要缺失的区域可以是具有基因编码区的总长度的10%或更多、20%或更多、30%或更多、40%或更多、50%或更多、60%或更多、70%或更多、80%或更多、90%或更多或95%或更多的长度的区域。此外,优选的是,要缺失的区域的上游和下游的序列的读码框不相同。读码框的不一致性可以引起要缺失的区域下游的移码。例如,通过将氨基酸取代(错义突变)、终止密码子(无义突变)、一个或两个核苷酸残基的添加或缺失(移码突变)的突变等引入染色体上的基因编码区中实现基因的破坏(journalofbiologicalchemistry,272:8611-8617(1997);proceedingsofthenationalacademyofsciences,usa,955511-5515(1998);journalofbiologicalchemistry,26116,20833-20839(1991))。也可以通过例如将另一个核苷酸序列插入染色体上基因的编码区来实现基因的破坏。插入位点可以位于基因的任何区域中,并且较长的核苷酸序列的插入通常可以更确定地使基因失活。优选的是,插入位点上游和下游的序列的读码框不相同。读码框的不一致性可以导致要缺失的区域下游的移码。其他核苷酸序列没有特别限制,只要选择降低或消除编码蛋白质活性的序列,并且其实例包括例如标志物基因如抗生素抗性基因和可用于产生目标物质的基因。特别地,可以以缺失编码蛋白质的氨基酸序列的方式进行基因的破坏。换言之,可以通过例如缺失蛋白质的氨基酸序列,特别地以编码氨基酸序列被缺失的蛋白质的方式修饰基因来实现降低蛋白质活性的修饰。术语“蛋白质的氨基酸序列的缺失”指蛋白质的氨基酸序列的部分或整个区域的缺失。另外,术语“蛋白质的氨基酸序列的缺失”指原始氨基酸序列在蛋白质中消失,并且也包括原始氨基酸序列改变为另一种氨基酸序列的情况。也就是说,例如,通过移码改变为另一种氨基酸序列的区域可以视为缺失区域。当缺失蛋白质的氨基酸序列时,通常缩短蛋白质的总长度,但也可以存在蛋白质的总长度没有改变或延长的情况。例如,通过缺失基因编码区的部分或整个区域,可以在编码的蛋白质中缺失由缺失区域编码的区域。另外,例如,通过将终止密码子引入基因的编码区中,可以在编码的蛋白质中缺失由引入位点的下游区域编码的区域。另外,例如,通过在基因的编码区中移码,可以在编码的蛋白质中缺失由移码区编码的区域。关于基因缺失中待缺失区域的位置和长度的上述描述可以在必要的变更后应用于缺失蛋白质氨基酸序列时待缺失区域的位置和长度。如上所述对染色体上的基因的此类修饰可以如下实现:通过例如制备以它不能产生正常起作用的蛋白质的方式进行修饰的破坏型基因并且用含有破坏型基因的重组dna转化宿主,以引起破坏型基因和染色体上的野生型基因之间的同源重组,从而用破坏型基因替换染色体上的野生型基因。在此程序中,若在重组dna中包含根据宿主特征选择的标志物基因如营养缺陷型,则实验操作变得更容易。破坏型基因的实例包括编码区的部分或整个区域被缺失的基因、包含错义突变的基因、包含无义突变的基因、包含移码突变的基因和包含转座子或标志物基因插入的基因。由破坏型基因编码的蛋白质即使得以产生也具有与野生型蛋白质的构象不同的构象,因此其功能得以降低或消除。已经建立了基于利用同源重组的基因取代的此类基因破坏,并且存在使用线性dna的方法,例如称为“red驱动整合”的方法(datsenko,k.a,andwanner,b.l.,proc.natl.acad.sci.usa,97:6640-6645(2000)),以及利用red驱动整合与源自λ噬菌体的切除系统组合的方法(cho,e.h.,gumport,r.i.,gardner,j.f.,j.bacteriol.,184:5200-5203(2002))(参见wo2005/010175)、使用具有温度敏感性复制起点的质粒的方法、使用能够接合转移的质粒的方法、利用不具有在宿主中起作用的复制起点的自杀载体的方法(美国专利no.6,303,383,日本专利公开(kokai)no.05-007491)等。用于降低蛋白质活性的修饰也可以通过例如诱变处理来实现。诱变处理的实例包括x射线或紫外线的照射和用诸如n-甲基-n’-硝基-n-亚硝基胍(mnng)、甲磺酸乙酯(ems)和甲磺酸甲酯(mms)的突变剂处理。如上所述的此类降低蛋白质活性的方法可以独立使用或以任意组合使用。当蛋白质作为由多个亚单位组成的复合物起作用时,可以修饰多个亚单位的部分或全部,只要蛋白质的活性最终得以降低即可。也就是说,例如,编码各个亚单位的多个基因的部分或全部可以得以破坏等。此外,当存在多种蛋白质的同工酶时,可以降低多种同工酶的部分或全部活性,只要蛋白质的活性最终得以降低即可。也就是说,例如,编码各个同工酶的多个基因的部分或全部可以得以破坏等。可以通过测量蛋白质的活性确认蛋白质活性的降低。也可以通过确认编码蛋白质的基因的表达降低来确认蛋白质活性的降低。可以通过确认基因转录量的减少或从基因表达的蛋白质量的减少来确认基因表达的降低。可以通过比较从基因转录的mrna量与未经修饰的菌株的mrna的量来确认基因转录量的减少。用于评估mrna量的方法的实例包括northern杂交、rt-pcr、微阵列、rna-seq等(molecularcloning,coldspringharborlaboratorypress,coldspringharbor(usa),2001)。优选地,mrna的量(例如每个细胞的mrna分子数)降低至例如未修饰菌株的50%或更小、20%或更小、10%或更小、5%或更小、或0%。蛋白质量的减少可以通过使用抗体的western印迹法来确认(molecularcloning,coldspringharborlaboratorypress,coldspringharbor(usa)2001)。优选地,蛋白质量(例如每个细胞的蛋白质分子数目)降低至例如未修饰菌株的50%或更小、20%或更小、10%或更小、5%或更小、或0%。可以根据破坏所用的方法,通过确定基因的部分或全部的核苷酸序列、限制酶图谱、全长等等来确认基因的破坏。上述降低蛋白质活性的方法可以适用于任意蛋白质的活性的降低和任意基因的表达的降低,以及核糖核酸酶iii活性的降低。<2>本发明的方法通过使用由此获得的本发明的细菌可以生产目标rna。本发明的方法是生产目标rna的方法,该方法包括培养本发明的细菌,并收集转录的目标rna。通过在培养基中培养本发明的细菌,可以将目标rna转录并在细菌的细胞中积累。也就是说,具体地,本发明的方法是用于生产目标rna的方法,该方法包括在培养基中培养本发明的细菌,转录目标rna并在细菌的细胞中积累目标rna,并从细胞中收集目标rna。本发明的细菌可以根据例如通常用于培养细菌如棒状杆菌细菌的培养条件进行培养。本发明的细菌可以在例如含有碳源、氮源和无机离子的常规培养基中培养。此外,例如,也可以根据需要添加有机微量营养素,例如维生素和氨基酸。作为碳源,可以使用碳水化合物如葡萄糖和蔗糖、有机酸如乙酸、醇等。作为氮源,例如,可以使用氨气、氨水、铵盐等。作为无机离子,例如,可以根据需要适当地使用钙离子、镁离子、磷酸根离子、钾离子、铁离子等。培养可以在ph5.0至8.5和15至37℃的适当范围内在需氧条件下进行10至120小时。此外,可以参考使用棒状杆菌等细菌生产l-氨基酸的培养条件和使用棒状杆菌细菌等细菌分泌生产蛋白质的方法的培养条件(wo01/23591,wo2005/103278,wo2013/065869,wo2013/065772,wo2013/118544,wo2013/062029等)。此外,当诱导型启动子用于表达目标rna时,可以适当地诱导目标rna的表达。通过在此类条件下培养本发明的细菌,将目标rna转录并在细菌的细胞中累积。目标rna的表达和积累可以通过例如将含有细胞提取物的级分作为样品应用于电泳,并检测对应于目标rna的分子量的条带来确认。可以通过用于分离和纯化化合物的适当方法从细胞中收集目标rna。此类方法的实例包括例如离心、盐析、乙醇沉淀、超滤、凝胶过滤层析、离子交换层析、亲和层析和电泳。这些方法可以独立使用,或者可以可以以适当组合使用。具体地,例如,可以用超声波等破坏细胞,可以通过离心等从细胞破碎的悬浮液中除去细胞获得上清液,并且可以通过离子交换树脂法等从上清液中收集目标rna。收集的目标rna可以是游离化合物、其盐或其混合物。另外,收集的目标rna也可以是具有高分子量化合物如蛋白质的复合物。也就是说,在本发明中,术语“目标rna”可以指游离形式的目标rna、其盐、其与高分子量化合物如蛋白质的复合物或其混合物,除非另有说明。盐的实例包括例如铵盐和钠盐。除了目标rna之外,收集的目标rna可以包含例如细菌细胞、培养基成分、水分和细菌的副产物代谢物等成分。还可以以期望的程度纯化目标rna。收集的目标rna的纯度可以是例如30%(w/w)或更高、50%(w/w)或更高、70%(w/w)或更高、80%(w/w)或更高、90%(w/w)或更高、或95%(w/w)或更高。实施例在下文中,将参考实施例更具体地解释本发明。然而,本发明不受这些实施例的限制。<1>获得谷氨酸棒杆菌的核糖核酸酶iii基因缺陷菌株以下述方式构建谷氨酸棒杆菌2256菌株(atcc13869菌株,以下也简称为“2256菌株”)的核糖核酸酶iii(rna酶iii)同源基因(以下也称为“rnc基因”)的破坏菌株。首先,基于与已知的rna酶iii的氨基酸序列同源性,将位于基因数据库(genbank)中谷氨酸棒杆菌2256菌株(登录号ap017557)的基因组序列信息的region:2115207..2115950的区域推断为rnc基因。然后,作为用于缺失该基因的必要信息,从基因数据库(genbank)获得其orf(读码框)区域和其上游和下游区域各约1000个核苷酸(1kb)的dna核苷酸序列信息。接下来,用dneasyblood&tissuekit(qiagen)从2256菌株的细胞获得基因组dna。通过使用该基因组dna作为模板和primestargxldna聚合酶(takarabio)以及seqidno:1和2的引物进行pcr扩增,以获得含有rnc上游区域的约1kb的dna片段,以及用seqidno:3和4的引物进行pcr扩增,以获得含有rnc基因下游区域的约1kb的dna片段。根据制造商推荐的方案设定pcr条件。然后,以下列方式将这些dna片段与含有sacb基因的质粒pbs4s(wo2005/113745和wo2005/113744;在谷氨酸棒杆菌中不具有复制能力)连接。具体地,通过使用pbs4s作为模板,seqidno:5和6的引物以及primestargxldna聚合酶进行pcr扩增,以获得pbs4s的扩增片段。然后,将上文获得的rnc基因的上游和下游区域的dna片段两者和pbs4s的扩增片段混合,并使用in-fusionhd克隆试剂盒(clontech)将这三个片段相互连接(图1)。用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,应用到含有25μg/ml卡那霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从琼脂培养基上出现的菌落中分离单菌落,以获得对卡那霉素具有抗性的转化体。以常规方式从获得的转化体中提取质粒。通过结构分析鉴定含有rnc基因上游和下游区域的dna片段的质粒,并命名为pbs4sδrnc(图1)。该质粒不能在棒状杆菌细菌中自主复制。因此,若用该质粒转化棒状杆菌细菌,则出现通过同源重组将该质粒掺入染色体并因此具有卡那霉素抗性的转化体,尽管它以极低的频率发生。因此,通过电脉冲方法用高浓度的质粒pbs4δrnc转化2256菌株,将其应用于含有25μg/ml卡那霉素的cm-dex琼脂培养基(5g/l葡萄糖,10g/l聚胨,10g/l酵母提取物,1g/lkh2po4,0.4g/lmgso4-7h2o,0.01g/lfeso4-7h2o,0.01g/lmnso4-7h2o,3g/l尿素,1.2g/l大豆水解物,用koh调节至ph7.5,20g/l琼脂),并在30℃培养过夜。结果,出现了几个菌落。在培养基上培养的这些菌株各自是所称作的一次重组菌株,其中由于质粒上的rnc基因附近(上游或下游)的dna序列片段和2256菌株基因组上rnc基因附近的区域之间的同源重组而将源自该质粒的卡那霉素抗性基因和sacb基因整合到基因组中。然后,将这些一次重组菌株各自在不含卡那霉素的cm-dex培养基(具有与cm-dex琼脂培养基相同的组成,只是它不含琼脂)中在30℃培养过夜。将培养液适当稀释,应用到不含卡那霉素的含10%(w/v)-蔗糖的dex-s10琼脂培养基(100g/l蔗糖,10g/l聚胨,10g/l酵母提取物,1g/lkh2po4,0.4g/lmgso4-7h2o,0.01g/lfeso4-7h2o,0.01g/lmnso4-4-5h2o,3g/l尿素,1.2g/l大豆蛋白水解物溶液,10μg/l生物素,用koh调节至ph7.5,20g/l琼脂),并在30℃培养过夜。结果,出现了几个菌落。这些菌株各自被认为是由于第二次同源重组而除去sacb基因并因此对蔗糖不敏感的菌株。由此获得的菌株将包括rnc基因被缺陷型替换的菌株,以及rnc基因恢复为野生型的菌株。因此,使用kodfxneo(toyobo)将出现的菌落应用于菌落pcr,以选择rnc基因缺陷菌株。由于通过用seqidno:7和8的引物进行pcr扩增分析了那些菌株的rnc基因区域的长度,一些菌株提供了pcr扩增长度比使用2256菌株(野生型)的基因组dna作为模板的情况观察到的长度短的dna片段。因此,选择其一个菌株作为rnc基因缺陷菌株,并命名为2256δrnc。顺便提及,对于野生型rnc基因的情况观察到的pcr扩增的dna片段的长度是约3kbp,而对于缺陷型rnc基因的情况观察到的pcr扩增的dna片段的长度是约2kbp(图2)。<2>内源性质粒的消除2256菌株具有内源质粒pam330(yamaguchi,ryuji,etal."determinationofthecompletenucleotidesequenceofbrevibacteriumlactofermentumplasmidpam330andanalysisofitsgeneticinformation."agriculturalandbiologicalchemistry50.11(1986):2771-2778.)。构建了质粒pvc7-sacb,其对应于掺入了sacb基因的质粒pvc7(jp1997-070291a)。pvc7是pam330和大肠杆菌通用载体phsg399(takarabio)的复合质粒。具体地,通过使用pbs4s作为模板,seqidno:9和10的引物以及primestargxldna聚合酶进行pcr扩增,以获得sacb基因的扩增片段。分别地,通过使用pvc7作为模板,seqidno:11和12的引物以及kodfxneo(toyobo)进行pcr扩增,以获得pvc7的扩增片段。将这两个扩增的片段混合,并使用in-fusionhd克隆试剂盒(clontech)相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,应用到含有25μg/ml氯霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并命名为pvc7-sacb(图3)。通过电脉冲方法各自用pvc7-sacb导入菌株2256和2256δrnc,将其应用于含有5μg/ml氯霉素的cm-dex琼脂培养基,并在30℃培养过夜,以获得菌株2256/pvc7-sacb和2256δrnc/pvc7-sacb中每种的多个转化体。然后,从它们获得菌株2256δpam330/pvc7-sacb和2256δrncδpam330/pvc7-sacb,其中除去了内源质粒pam330。然后,将这些菌株各自应用到dex-s10琼脂培养基,并在30℃培养过夜,以获得菌株2256δpam330和2256δrncδpam330,其被消除pvc7-sacb,从而变得对蔗糖不敏感。<3>单向转录质粒pvc7-pf1-u1ainsert的构建以下述方式构建质粒pvc7-pf1-u1ainsert,其用于在单一方向上在f1启动子的控制下转录作为目标rna的u1a结合序列。选择来自噬菌体bfk20(其对棒状杆菌细菌具有感染性)的启动子序列(登录号l13772)的启动子功能区(seqidno:13;也称为“f1启动子”;koptides,m.,etal.,(1992).characterizationofbacteriophagebfk20frombrevibacteriumflavum.microbiology,138(7),1387-1391.)、其终止子(ter)区域(seqidno:14;bukovska,g.,etal.,(2006).completenucleotidesequenceandgenomeanalysisofbacteriophagebfk20-alyticphageoftheindustrialproducerbrevibacteriumflavum.virology,348(1),57-71.)和u1a结合序列作为目标rna(seqidno:15,其中“t”应当读作“u”;endoh,t.,etal.,(2008).cellularsirnadeliverymediatedbyacell-permeantrna-bindingproteinandphotoinducedrnainterference.bioconjugatechemistry,19(5),1017-1024.)(其具有对小核仁核糖核蛋白u1a的结合能力),并且设计了u1a结合序列的转录单元的dna序列(u1ainsertrna转录单元;seqidno:16)(图4)。通过化学合成(eurofinsgenomics)制备u1ainsertrna转录单元的dna片段。然后,通过使用dna片段作为模板,seqidno:17和18的引物,以及kodfxneo(toyobo)进行pcr扩增,以获得u1ainsertrna转录单元的扩增片段。分别地,通过使用pvc7作为模板,seqidno:19和20的引物以及kodfxneo(toyobo)进行pcr扩增,以获得pvc7的扩增片段。将这些扩增的片段混合,并通过使用in-fusionhd克隆试剂盒(clontech)相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,应用到含有25μg/ml氯霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落以获得转化体。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为pvc7-pf1-u1ainsert(图4)。<4>双向转录质粒pvc7-pf1-hv-iap-pf1rev的构建以下述方式构建质粒pvc7-pf1-hv-iap-pf1rev,其用于在双重方向上在f1启动子的控制下转录作为目标rna的hv-iaprna。根据wo2010/140675中描述的信息,通过化学合成制备hv-iap的dna片段(seqidno:21),其是编码茄二十八星瓢虫(henosepilachnavigintioctopunctata)的凋亡蛋白抑制剂iap的iap基因的cdna的部分序列。以下述方式构建含有dna序列的质粒,所述dna序列含有f1启动子以及其直接下游连接的hv-iap序列(图5a)。首先,通过使用pvc7作为模板,seqidno:20和22的引物,以及kodfxneo(toyobo)进行pcr扩增,以获得pvc7的扩增片段。分别地,通过使用seqidno:16的dna片段作为模板,seqidno:23和24的引物,以及primestarhs(takarabio)进行pcr扩增,以获得含有f1启动子序列的dna片段。分别地,通过使用seqidno:21的dna片段作为模板,seqidno:25和26的引物,以及primestarhs(takarabio)进行pcr扩增,以获得hv-iap序列的dna片段。将这三个dna片段混合,并使用in-fusionhd克隆试剂盒(clontech)相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,并且获得对25μg/ml氯霉素具有抗性的菌株。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将它们之一命名为pvc7-pf1-hv-iap(图5a)。通过使用pvc7作为模板,seqidno:20和27的引物以及kodfxneo(toyobo)进行pcr扩增,以获得pvc7的扩增片段。分别地,通过使用seqidno:16的dna片段作为模板,seqidno:28和29的引物和primestarhs(takarabio)进行pcr扩增,以获得f1启动子序列的扩增片段。将这两个扩增的片段混合,并使用in-fusionhd克隆试剂盒(clontech)相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,应用到含有25μg/ml氯霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为pvc7-pf1rev(图5b)。接下来,为了在pvc7-pf1rev的f1启动子下游导入限制性酶切位点kpni和xhoi,通过使用pvc7-pf1rev作为模板,seqidno:20和30的引物,以及kod-plus-诱变试剂盒(toyobo)进行反向pcr。然后,将扩增的dna片段进行dpni处理,磷酸化反应和自连接反应,从而环化,并导入大肠杆菌jm109菌株(takarabio)的感受态细胞中。将细胞应用于含有25μg/ml氯霉素的lb琼脂培养基,并在37℃培养过夜。然后,从出现的菌落中分离单菌落。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为pvc7-kpni-xhoi-pf1rev(图5b)。然后,通过使用pvc7-pf1-hv-iap作为模板,seqidno:31和32的引物,和primestarhs(takarabio)进行pcr,以获得依次含有kpni限制酶位点、f1启动子区、hv-iap区域和xhoi限制酶位点的dna片段。用限制酶kpni和xhoi各自消化此dna片段和pvc7-kpni-xhoi-pf1rev,并用minelutepcr纯化试剂盒(qiagen)纯化。将这两种纯化的产物混合,并通过使用ligationhighver.2(toyobo)的连接反应相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,并且获得对25μg/ml氯霉素具有抗性的菌株。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为pvc7-pf1-hv-iap-pf1rev(图5b)。<5>目标rna的产生通过电脉冲方法将质粒pvc7、pvc7-pf1-u1ainsert和pvc7-pf1-hv-iap-pf1rev各自导入到谷氨酸棒杆菌菌株2256δpam330和2256δrncδpam330中,将所述菌株应用于含有5μg/ml氯霉素的cm-dex琼脂培养基,并在30℃培养过夜。因此,获得转化体菌株2256δpam330/pvc7和2256δrncδpam330/pvc7,2256δpam330/pvc7-pf1-u1ainsert和2256δrncδpam330/pvc7-pf1-u1ainsert,和2256δpam330/pvc7-pf1-hv-iap-pf1rev和2256δrncδpam330/pvc7-pf1-hv-iap-pf1rev。将上文获得的转化体的菌落各自涂布在含有5μg/ml氯霉素的cm-dex琼脂培养基上,并在30℃培养约16小时。然后,将培养细胞的部分用于试管培养。在2ml含有5μg/ml氯霉素的cm-dex培养基中于30℃振荡培养24小时。然后,用rnaprotectbacteriareagent处理200μl培养液,并且取出上清液。然后,向其中添加225μl含有15mg/ml溶菌酶(sigma)的te缓冲液以在室温进行反应30分钟,进一步向其中添加25μl的20mg/mlprok(takarabio)以在室温进行反应30分钟,然后用trizolls(thermofisherscientific)提取rna。将提取的rna用50μl不含rna酶的水溶解以制备总rna溶液。使用novextbegels,6%(thermofisherscientific)对获得的总rna溶液进行总rna分析。也就是说,将总rna溶液各1μl应用于凝胶泳道,并在非变性条件下进行聚丙烯酰胺凝胶电泳(page)。因此,当比较从菌株2256δpam330/pvc7-pf1-u1ainsert和2256δrncδpam330/pvc7-pf1-u1ainsert中提取的总rna时,仅在rnc基因缺陷的菌株的预测位置处确认源自u1ainsert的rna条带(图6)。类似地,当比较从菌株2256δpam330/pvc7-pf1-hv-iap-pf1rev和2256δrncδpam330/pvc7-pf1-hv-iap-pf1rev中提取的总rna时,也仅在rnc基因缺陷的菌株的预测位置处确认源自hv-iap的rna条带(图6)。因此,显示rnc基因的缺失对使用棒状杆菌属细菌的rna生产是有效的。<6>获得高拷贝数变异质粒将转化体2256/pvc7的60个克隆各自在含有5μg/ml氯霉素的cm-dex培养基中培养过夜。将500μl等分试样的培养液离心(14,400xg,2分钟),并给收集的细胞添加200μl溶于p1缓冲液(qiagen)中的来自鸡蛋清(sigma-aldrich)的15mg/ml溶菌酶在37℃进行反应30分钟。然后,用qiaprepspinminiprepkit(qiagen)从每个克隆的细胞中提取质粒,并进行琼脂糖凝胶电泳以比较质粒量。因此,观察到从克隆之一提取的质粒提供了明显比从其他克隆中提取的dna带更粗大的dna带。因此,将此质粒重新导入2256菌株中,类似地从转化体中提取质粒,并通过琼脂糖凝胶电泳分析其产量。如上所述,此质粒提供了明显比作为对照的pvc7的dna带更粗大的dna带,也就是说,揭示了此质粒是与原始质粒pvc7相比在棒状杆菌细菌细胞中以高拷贝数维持的质粒。将此质粒命名为pvc7h1。用dna测序仪geneticanalyzer3500xl(appliedbiosystems)分析获得的pvc7h1的突变位点。因此,揭示了在pvc7h1中,pvc7的6679bp总核苷酸序列的第1172核苷酸已经从胞嘧啶(c)突变为腺嘌呤(a),其中从限制酶hindiii的消化识别位点的5’末端计数的第2位的核苷酸a视为“+1”。以下,此突变称为“c1172a”。因此,揭示了此突变是pvc7h1的高拷贝数变异的原因。因为揭示了c1172a突变显著增加pvc7的拷贝数,所以将表1中所示的此区域处的突变各自导入pvc7中,并评估这些突变对质粒拷贝数的影响。用kod-plus-诱变试剂盒(toyobo)构建突变体质粒。根据试剂盒所附的构建方案(表1),与作为模板的质粒pvc7组合,通过使用seqidno:33和34的引物构建pvc7h2,通过使用seqidno:33和35的引物构建pvc7h3,通过使用seqidno:36和37的引物构建pvc7h4,通过使用seqidno:36和38的引物构建pvc7h5,通过使用seqidno:33和39的引物构建pvc7h6,以及通过使用seqidno:33和40的引物构建pvc7h7。用dna测序仪确认其总核苷酸序列是正确的。[表1]表1:pvc7突变体质粒在核苷酸序列中,与野生型的核苷酸不同的核苷酸加下划线,并且缺失位点以“*”表示。以常规方式将构建的质粒各自转化到2256δpam330菌株中。含有5μg/ml氯霉素的cm-dex琼脂培养基用作选择培养基。在转化任何质粒的情况下,菌落是良好地形成的。因此,用接种环将含有相应质粒的菌落各自接种到相同琼脂培养基中,并在30℃培养约1天。然后,将每个菌株的生长细胞以约2平方厘米的量接种到含有5μg/ml氯霉素的cm-dex培养基中,并在30℃振荡培养过夜。以常规方式从每种培养液中提取含有的质粒。将制备的质粒溶液的部分进行琼脂糖凝胶电泳以分析每种质粒的dna条带。表2中显示了结果。[表2]表2:电泳图上的质粒的条带强度no.质粒名称dna条带的强度*11pvc7h2++++++2pvc7h3++++3pvc7h4++++++4pvc7h5+++++5pvc7h6++++6pvc7h7+++7pvc7h1++++8pvc7+*1:从含有质粒的细胞中提取每种质粒,并且通过琼脂糖凝胶电泳中的视觉观察评估其dna条带的强度。“+”的较大数目指示质粒的较大产率量。因此,获得了突变体质粒,其拷贝数明显高于最初获得的pvc7h1的拷贝数。使用它们之一,即pvc7h2进行以下实验。估计pvc7的拷贝数为约10个拷贝/细胞,pvc7h1的拷贝数为约100个拷贝/细胞,并且pvc7h2的拷贝数为约250个拷贝/细胞。<7>使用高拷贝数变异质粒的rna生产以下述方式构建pvc7-pf1-u1ainsert和pvc7-pf1-hv-iap-pflrev的高拷贝数变异。具体地,通过使用pvc7-pf1-u1ainsert或pvc7-pf1-hv-iap-pf1rev作为模板、用于引入pvc7h1突变的seqidno:33和41的引物或用于引入pvc7h2突变的seqidno:33和34的引物和kod-plus-诱变试剂盒(toyobo)进行反向pcr。然后,将获得的dna片段进行dpni处理,磷酸化反应和自连接反应,从而环化,并转化到大肠杆菌jm109菌株(takarabio)的感受态细胞中。将细胞应用于含有25μg/ml氯霉素的lb琼脂培养基,并在37℃培养过夜。然后,从出现的菌落中分离单菌落以获得转化体。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为pvc7h1-pf1-u1ainsert、pvc7h2-pf1-u1ainsert、pvc7h1-pfl-hv-iap-pflrev和pvc7h2-pf1-hv-iap-pflrev。接下来,通过电脉冲法给2256δrncδpam330菌株导入质粒pvc7、pvc7-pf1-u1ainsert、pvc7h1-pf1-u1ainsert、pvc7h2-pf1-u1ainsert、pvc7-pf1-hv-iap-pf1rev、pvc7h1-pf1-hv-iap-pf1rev和pvc7h2-pf1-hv-iap-pf1rev中的每种,应用于含有5μg/ml氯霉素的cm-dex琼脂培养基,并在30℃培养过夜。由此,获得转化体菌株2256δrncδpam330/pvc7,2256δrncδpam330/pvc7-pf1-u1ainsert,2256δrncδpam330/pvc7h1-pf1-u1ainsert,2256δrncδpam330/pvc7h2-pf1-u1ainsert,2256δrncδpam330/pvc7-pf1-hv-iap-pf1rev,2256δrncδpam330/pvc7h1-pf1-hv-iap-pf1rev和2256δrncδpam330/pvc7h2-pf1-hv-iap-pf1rev。然后,将这些菌株各自在试管中培养,并且评估由菌株产生的rna。将来自每个转化体的菌落的琼脂培养基上培养的细胞的部分接种到2ml含有5μg/ml氯霉素的cm-dex培养基中,并在30℃振荡培养24小时。然后,以与上述实施例相同的方式,包括rnaprotectbacteriareagent处理从200μl培养液中提取rna,最后用50μl不含rna酶的水溶解rna样品以制备总rna溶液。使用novextbe凝胶(6%)在非变性条件下通过page对获得的总rna溶液进行总rna分析。因此,u1ainsert-rna的累积量以下述顺序增加:pvc7-pf1-u1ainsert<<pvc7h1-pf1-u1ainsert,约等于pvc7h2-pf1-u1ainsert(图7)。类似地,hv-iap-dsrna的累积量以下述顺序增加:pvc7-pf1-hv-iap-pf1rev<<<pvc7h1-pf1-hv-iap-pf1rev<pvc7h2-pf1-hv-iap-pf1rev(图7)。因此,显示通过增加用于rna转录的质粒的拷贝数来增加目标rna的细胞内累积量。<8>与大肠杆菌中由t7-启动子诱导表达系统的rna生产的比较如前所述(timmons,l.,court,d.l.,&fire,a.(2001).ingestionofbacteriallyexpresseddsrnascanproducespecificandpotentgeneticinterferenceincaenorhabditiselegans.gene,263(1),103-112.),已经报道了在大肠杆菌ht115(de3)菌株中使用t7rna聚合酶的rna生产系统,所述菌株是rnc基因缺陷菌株。因此,在谷氨酸棒杆菌2256δrncδpam330菌株中由f1-启动子表达系统的rna生产和大肠杆菌中由t7-启动子诱导表达系统的rna生产之间进行比较。为此,以下述方式构建用于在单一方向上在t7启动子控制下转录u1a结合序列的质粒pl4440-pt7-u1ainsert和用于在双重方向上在t7启动子控制下转录hv-iaprna的质粒pl4440-pt7-hv-iap-pt7rev。通过使用质粒pl4440(gehealthcare)作为模板、seqidno:42和43的引物以及kodfxneo进行pcr,以获得pl4440的dna片段。分别地,通过使用pvc7-pf1-u1ainsert作为模板、seqidno:44和45的引物以及primestarhs(takarabio)进行pcr,以获得含有u1ainsert序列的dna片段。分别地,混合seqidno:46的dna链和seqidno:47的dna链,其中seqidno:47的dna链具有对应于seqidno:46的dna链的互补序列的序列,并且将它们相互退火,并用minelutepcr纯化试剂盒纯化,以获得t7终止子序列的dna片段。将这三个dna片段混合,并通过使用in-fusionhd克隆试剂盒相互连接。然后,用反应混合物转化大肠杆菌jm109菌株的感受态细胞,应用到含有100μg/ml氨苄青霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将它们之一命名为pl4440-pt7-u1ainsert(图8)。通过使用质粒pl4440作为模板、seqidno:42和48的引物以及kodfxneo进行pcr,以获得pl4440的dna片段。分别地,通过使用质粒pvc7-pt7-hv-iap-pt7rev(下文实施例9(2-2)中所述)作为模板、seqidno:49和50的引物以及kodfxneo进行pcr,以获得“pt7-hv-iap-pt7rev”区域的dna片段。将这些dna片段混合,并通过使用in-fusionhd克隆试剂盒相互连接。然后,用反应混合物转化大肠杆菌jm109菌株的感受态细胞,应用到含有100μg/ml氨苄青霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落以获得转化体。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将它们之一命名为pl4440-pt7-hv-iap-pt7rev(图9)。通过电脉冲法给大肠杆菌ht115(de3)菌株导入pl4440-pt7-u1ainsert和pl4440-pt7-hv-iap-pt7rev中的每种,以获得转化株菌株ht115(de3)/pl4440-pt7-u1ainsert和ht115(de3)/pl4440-pt7-hv-iap-pt7rev。将菌株各自在试管中含有100μg/ml氨苄青霉素的lb液体培养基中于37℃振荡培养过夜以制备种子培养液。接下来,将种子培养液以培养基的1/50的体积添加到含有100μg/ml氨苄青霉素的lb液体培养基中,以在37℃开始主培养。在振荡培养3小时后,向其中添加终浓度1mm的iptg,再继续培养,并在添加iptg后4小时、8小时和24小时进行取样。将200μl等分试样的培养液离心(13,800xg,2分钟)以收集细胞。用trizol(thermofisherscientific)根据其方案提取rna,并用50μl不含rna酶的水溶解以制备总rna溶液。为了比较,使用实施例<7>中获得的谷氨酸棒杆菌的总rna溶液。通过使用novextbe凝胶(6%)和1μl每种rna样品在非变性条件下进行page。因此,谷氨酸棒杆菌中由f1启动子表达系统产生的u1ainsert-ssrna的量大于大肠杆菌中由t7启动子诱导表达系统产生的u1ainsert-ssrna的量(图10)。类似地,谷氨酸棒杆菌中由f1启动子表达系统产生的hv-iap-dsrna的量大于大肠杆菌中由t7启动子诱导表达系统产生的hv-iap-dsrna的量(图10)。因此,揭示了诸如谷氨酸棒杆菌的棒状杆菌细菌可用于产生目标rna。<9>谷氨酸棒杆菌中由t7-启动子诱导的表达系统的rna生产(1)发夹样结构rna的生产(1-1)质粒ppk-t7lac-vd-antiolac的构建以转录发夹样结构rna以下述方式构建质粒ppk-t7lac-vd-antiolac,其用于在单一方向上在t7启动子的控制下转录发夹样结构rna作为目标rna。(1-1-1)puc57-vd的构建通过化学合成基于马铃薯纺锤体类病毒(pstvd).125(molbiol(mosk).2013jan-feb;47(1):94-106.)的基因组序列制备编码发夹样结构rna的seqidno:53的dna片段。将dna片段克隆到puc57(atgservis-gen,st.petersburg,russia)中以构建puc57-vd。(1-1-2)ppk-t7lac的构建首先,通过pcr用seqidno:54和55的引物从pet22b(+)(novagen)扩增含有终止子t7(tt7)的片段(131bp)。然后,用kpni和sali消化该片段,并克隆到用相同的限制酶线性化的ppk4(美国专利号6,090,597)中。将构建的质粒命名为ppk4xb-t7ter(图11)。然后,通过pcr用seqidno:56和57的引物从pet22b(+)扩增含有laci-pt7-olac-mcs-tt7的dna区域的另一个片段(1959bp)。用kpni消化扩增的片段,并克隆到用kpni线性化的ppk4xb-t7ter中。构建的质粒命名为ppk-t7lac(图12)。(1-1-3)ppk-t7lac-vd-antiolac的构建用xbai和xhoi从puc57-vd切下dna片段vd-aolac,并用t4dna连接酶克隆到ppk-t7lac中。通过电穿孔将连接混合物转化到大肠杆菌le392菌株(promega)的感受态细胞中。应用大肠杆菌细胞的标准电穿孔程序。细胞在37℃生长1.5小时后,将细胞接种在补充有50μg/ml卡那霉素(km)的lb琼脂培养基平板上,以获得卡那霉素抗性(kmr)转化体。从转化体中提取质粒,选择目的质粒并命名为ppk-t7lac-vd-antiolac(图13)。(1-2)用于表达t7rna聚合酶的质粒pvc54-t7pol的构建以下述方式构建用于表达t7rna聚合酶的质粒pvc54-t7pol。(1-2-1)pvc54的构建对于pvc54的构建,以下述方式将含有氯霉素抗性(cmr)基因的启动子区和源自pvc7的pam330的部分(美国专利号5,804,414)的seqidno:58的区域替换为含有强启动子的区域。通过化学合成制备含有强启动子的seqidno:58的dna片段-a(161bp)。分别地,使用seqidno:60(l54-cm)和61(r55-cm)的引物,以pvc7质粒为模板,通过pcr扩增dna片段-b。通过使用片段-a和片段-b进行重叠-pcr,并使用seqidno:62(l54-hpa)和61(r55-cm)的侧翼引物通过pcr再次扩增得到的pcr片段。然后,用hpai和ncoi消化扩增的片段,并连接到pvc7上的hpai-ncoi位点。将连接混合物转化到大肠杆菌tg1菌株(zymoresearch)的感受态细胞中以获得cmr转化体。从转化体中提取质粒,选择目的质粒并命名为pvc54(图14)。(1-2-2)pbs5t-ptrb*-t7pol的构建使用seqidno:63(x550)和64(x551)的引物,用2256菌株的基因组dna作为模板,通过pcr扩增含有ptrb和hemh基因的部分的dna片段(2021bp)。将扩增的片段用bglii消化,并克隆到pbs5t(wo2006/057450)的bamhi位点。得到的质粒命名为pbs5t-ptrb*(图15)。它在ptrb基因部分中含有独特的nhei位点。分别地,以pah162-λattl-tcr-λattr(bmcbiotechnology2008,8:63)作为模板,使用seqidno:65(x553)和66(x554)的引物通过pcr扩增含有rrnb终止子(trrnb)和l3终止子(tl3)的另一个dna片段(531bp)。将扩增的片段用nhei消化,并克隆到pbs5t-ptrb*的nhei位点中。得到的质粒命名为pbs5t-ptrb*-2ter(图16)。它在终止子trrnb和tl3之间含有独特的bamhi位点,并且两个终止子侧翼都有bglii位点。最后,用bamhi从par1219(procnatlacadsciusa.1984apr;81(7):2035-2039)切下含有laci-placuv5-gene1的dna片段(4452bp),并克隆到pbs5t-ptrb*-2ter的独特bamhi位点中。laci-placuv5-gene1片段含有编码t7rna聚合酶的基因1,并在lacuv5启动子(placuv5)的控制下表达。选择具有gene1至tl3遗传方向的质粒并命名为pbs5t-ptrb*-t7pol(图17)。(1-2-3)pvc54-t7pol的构建以下述方式从pvc54和pbs5t-ptrb*-t7pol构建用于表达t7rna聚合酶的质粒pvc54-t7pol。首先,用bglii从pbs5t-ptrb*-t7pol中切出含有laci-placuv5-gene1的dna片段。然后,将dna片段在bamhi位点处连接到pvc54中以构建pvc54-t7pol(图18)。(1-3)生产rna的谷氨酸棒杆菌的构建最初通过电转化法用pvc54-t7pol转化谷氨酸棒杆菌2256δrncδpam330菌株。在具有补充有10μg/ml氯霉素(cm)的cm2g培养基(5g/l葡萄糖,10g/l胰蛋白胨,10g/l酵母提取物,5g/lnacl,调节至ph7.0)的平板上在30℃培养24小时选择转化体。然后,应用单菌落分离方法获得具有pvc54-t7pol的两个独立的谷氨酸棒杆菌2256δrncδpam330克隆的菌落。用ppk-t7lac-vd-antiolac对那两个克隆进行另一次转化。在具有补充有10μg/mlcm和25μg/mlkm的cm2g培养基的平板上在30℃生长24小时选择转化体。再次,应用单菌落分离方法获得具有pvc54-t7pol和ppk-t7lac-vd-antiolac的谷氨酸棒杆菌2256δrncδpam330的两个独立克隆(命名为克隆a和b)的菌落(命名为谷氨酸棒杆菌2256δrncδpam330/pvc54-t7pol/ppk-t7lac-vd-antiolac)。(1-4)谷氨酸棒杆菌中由t7启动子诱导表达系统的rna生产将谷氨酸棒杆菌2256δrncδpam330/pvc54-t7pol/ppk-t7lac-vd-antiolac菌株(克隆a和b中的每种)接种到5ml补充有10μg/mlcm和25μg/mlkm的cm2g培养基中,并在32℃培养过夜。次日早晨,将菌株接种到od600值为0.2-0.3的相同新鲜培养基(5ml)中,并在32℃培养4-5小时。此时,通过向培养液中添加2mmiptg进行诱导。诱导后,继续再培养3小时和19小时,并还测量培养液的od600值。然后,将每种培养液的等分试样(1-2ml)立即与rnaprotectbacteriareagent(qiagen76506)根据制造推荐混合,并立即将细胞团粒在-70℃冷冻。将所有样品各自在4℃融化,并使用trizolls(ambion10296028)进行rna分离程序。将分离的总rna在30μl经depc处理的水中稀释以制备总rna溶液,通过nanodrop分光光度计测量溶液中rna的浓度,然后将溶液的等分试样用于变性尿素-page(5%)。通常将3μg每种总rna溶液应用到凝胶孔中。电泳后,用溴化乙啶(ethbr)对凝胶中的rna进行染色。因此,对iptg诱导的样品观察到目标rna条带(vd相关的rna)(图19)。也就是说,确认了在谷氨酸棒杆菌中通过使用t7-启动子诱导的表达系统进行了rna生产。(2)hv-iaprna的生产(2-1)用于表达t7rna聚合酶的质粒ppk4-t7pol的构建以下述方式构建用于表达t7rna聚合酶的质粒ppk4-t7pol。使用kodfxneo聚合酶、seqidno:67和68的引物以及pvc54-t7pol作为模板,通过pcr扩增含有编码t7rna聚合酶的基因1(t7pol)的dna片段。使用seqidno:69和70的引物和质粒ppk4(美国专利号6,090,597)作为模板,通过pcr获得另一个dna片段。将这两个dna片段混合,并使用in-fusionhd克隆试剂盒(takarabio)相互连接。然后,用反应混合物转化大肠杆菌jm109感受态细胞,应用到含有km(50μg/ml)的lb琼脂培养基上,并在37℃培养16小时以获得kmr转化体。其中,分离了几个菌落,并且从转化体中提取质粒。在确认质粒的dna序列后,选择目的质粒并命名为ppk4-t7pol(图20)。(2-2)用于转录hv-iaprna的质粒pvc7-pt7-hv-iap-pt7rev的构建以下述方式构建质粒pvc7-pt7-hv-iap-pt7rev,其用于在双重方向上在t7启动子的控制下转录hv-iaprna以作为目标rna。(2-2-1)pvc7-pt7-kpni-xhoi-pt7rev的构建使用kodfxneo、seqidno:71和72的引物以及pvc7作为模板,通过pcr获得dna片段-n。另外,通过化学合成制备seqidno:73的dna片段-p和seqidno:74的另一个dna片段-q,所述dna片段-p依次含有t7启动子(正向)、kpni限制性位点、xhoi限制性位点和t7-启动子(反向),所述另一个dna片段-q含有片段-p的互补序列,并将这两个单链dna片段混合并退火以产生dna片段-r。然后,通过使用in-fusionhd克隆试剂盒将dna片段-n和dna片段-r两者彼此连接。用反应混合物转化大肠杆菌jm109感受态细胞,应用到含有cm(25μg/ml)的lb琼脂培养基上,并在37℃培养16小时以获得cmr转化体。其中,分离了几个菌落,并且从转化体中提取质粒。在确认质粒的dna序列后,选择目标质粒并命名为pvc7-pt7-kpni-xhoi-pt7rev(图21)。(2-2-2)pvc7-pt7-hv-iap-pt7rev的构建使用seqidno:75的dna片段作为模板和seqidno:76和77的引物,通过pcr扩增依次含有kpni限制性位点、hv-iap和xhoi限制性位点的dna片段-s。然后,dna片段-s和pvc7-pt7-kpni-xhoi-pt7rev各用kpni和xhoi消化,并用minelutepcr纯化试剂盒(qiagen)纯化。将这两种纯化的产物混合,并使用ligationhighver.2(toyobo)相互连接。用反应混合物转化大肠杆菌jm109感受态细胞,应用到含有cm(25μg/ml)的lb琼脂培养基上,并在37℃培养16小时以获得cmr转化体。其中,分离了几个菌落,并且从转化体中纯化质粒。在确认质粒的dna序列后,选择目的质粒并命名为pvc7-pt7-hv-iap-pt7rev(图21)。(2-3)生产rna的谷氨酸棒杆菌的构建通过电穿孔给谷氨酸棒杆菌2256δrncδpam330菌株导入ppk4-t7pol。将细胞悬浮液应用于含有km(25μg/ml)的cm-dex琼脂培养基,并在30℃培养16小时以获得转化体。随后,给转化体之一导入pvc7-pt7-hv-iap-pt7rev。将细胞悬浮液应用于含有km(25μg/ml)和cm(5μg/ml)的cm-dex琼脂培养基,并在30℃培养24小时以获得转化体。因此,最终获得谷氨酸棒杆菌2256δrncδpam330/ppk4-t7pol/pvc7-pt7-hv-iap-pt7rev。(2-4)谷氨酸棒杆菌中由t7启动子诱导表达系统的rna生产将谷氨酸棒杆菌2256δrncδpam330/ppk4-t7pol/pvc7-pt7-hv-iap-pt7rev菌株在30℃在含有含km(25μg/ml)和cm(5μg/ml)的cm-dex培养基的试管中培养16小时制备种子培养液。然后,将种子培养液以培养基体积的十分之一接种到新鲜cm-dex培养基中,以在30℃开始主培养。温育6小时后,对培养液的等分试样取样,并且对培养液的剩余部分添加2mmiptg,并进一步培养。在iptg添加后3小时和27小时,对培养液的等分试样取样。以与上述实施例相同的方式,包括rnaprotectbacteriareagent处理从200μl培养液中提取rna,最后用50μl不含rna酶的水溶解rna样品以制备总rna溶液。将总rna溶液各自应用于novextbe凝胶(6%),并分析rna生产。因此,对用iptg诱导后27小时获得的样品观察到目标rna带(hv-iaprna)(图22)。也就是说,再次证实了谷氨酸棒杆菌中通过使用t7-启动子诱导表达系统进行的rna生产。<10>ppk4的高拷贝数变异质粒的获得质粒ppk4是可用于大肠杆菌的质粒phsg399和谷氨酸棒杆菌atcc13058所具有的质粒phm1519的复合质粒,因此,ppk4充当可在这两种细菌中复制的穿梭载体(美国专利号6,090,597)。将表3中所示的突变分别导入ppk4,并且评估这些突变对质粒拷贝数的影响。用kod-plus-诱变试剂盒(toyobo)分别构建突变体质粒。与质粒ppk4作为模板组合,根据试剂盒附带的构建方案,通过使用seqidno:79和80的引物构建ppk4h1,通过使用seqidno:81和82的引物构建ppk4h2,通过使用seqidno:83和82的引物构建ppk4h3,通过使用seqidno:84和82的引物构建ppk4h4,通过使用seqidno:85和82的引物构建ppk4h5,并且通过使用seqidno:86和82的引物构建ppk4h6(表3)。用dna测序仪证实其总核苷酸序列是正确的。[表3]表3:ppk4突变体质粒在核苷酸序列中,与野生型的核苷酸不同的核苷酸是加下划线的。以与实施例<5>相同的方式分析构建的质粒的拷贝数。因此,揭示了表3中所示的所有ppk4突变体质粒在谷氨酸棒杆菌中显示比原始质粒ppk4更高的拷贝数。其中,ppk4h1显示最高的拷贝数,其在宿主的每个染色体上达到约200拷贝或更高。<11>使用ppk4h1的rna生产以下述方式将u1a-rna的表达系统整合到质粒ppk4h1中。通过使用pvc7-pf1-u1ainsert作为模板、seqidno:87和88的引物以及kodfxneo(toyobo)进行pcr扩增,以获得u1ainsertrna转录单元的扩增片段。分别地,通过使用ppk4h1作为模板、seqidno:89和90的引物以及kodfxneo(toyobo)进行pcr扩增,以获得ppk4h1的扩增片段。将这些扩增的片段混合,并使用in-fusionhd克隆试剂盒(clontech)相互连接。然后,用反应混合物转化大肠杆菌jm109菌株(takarabio)的感受态细胞,应用到含有50μg/ml卡那霉素的lb琼脂培养基上,并在37℃培养过夜。然后,从出现的菌落中分离单菌落以获得转化体。以常规方式从获得的转化体中提取质粒。通过dna测序分析鉴定目标质粒,并将其命名为ppk4h1-pf1-u1ainsert。通过电脉冲法将质粒ppk4h1和ppk4h1-pf1-u1ainsert中的每种导入到谷氨酸棒杆菌菌株2256δrncδpam330中,将其应用于含有25μg/ml卡那霉素的cm-dex琼脂培养基,并在30℃培养过夜。由此,获得转化体菌株2256δrncδpam330/ppk4h1和2256δrncδpam330/ppk4h1-pf1-u1ainsert。选择用于转化体菌株的各两个菌落,涂布在含有25μg/ml卡那霉素的cm-dex琼脂培养基上,并在30℃培养约16小时。然后,将培养细胞的部分用于试管培养。在2ml含有25μg/ml卡那霉素的cm-dex培养基中于30℃振荡培养24小时。然后,用rnaprotectbacteriareagent处理200μl培养液,并且除去上清液。然后,向其中添加225μl含有15mg/ml溶菌酶(sigma)的te缓冲液以在室温进行反应30分钟,进一步向其中添加25μl的20mg/mlprok(takarabio)以进行在室温反应30分钟,然后用trizolls(thermofisherscientific)提取rna。将提取的rna用50μl不含rna酶的水溶解以制备总rna溶液。使用novextbegels,6%(thermofisherscientific)对获得的总rna溶液进行总rna分析。也就是说,将各1μl总rna溶液应用于凝胶泳道,并在非变性条件下进行聚丙烯酰胺凝胶电泳(page)。因此,仅在菌株22256δrncδpam330/ppk4h1-pf1-u1ainsert的预测位置处确认了源自u1ainsert的rna条带(图23)。因此,即使当使用phm1519衍生的载体(其不同于前述实施例中使用的pvc7衍生的载体)时,在作为宿主的棒状杆菌属细菌中成功生产了大量的目标rna。工业适用性根据本发明,可以有效生产rna。<序列表的说明>seqidnos:1-12:引物13:的核苷酸序列f1启动子14:bfk20终止子区域的核苷酸序列15:u1a结合序列的核苷酸序列16:u1ainsertrna转录单元的的核苷酸序列17-20:引物21:hv-iap的核苷酸序列22-50:引物51:谷氨酸棒杆菌2256(atcc13869)的rnc基因的核苷酸序列52:谷氨酸棒杆菌2256(atcc13869)的rnc蛋白的氨基酸序列53:dna片段的核苷酸序列54-57:引物58:dna片段的核苷酸序列59:dna片段-a的核苷酸序列60-72:引物73:dna片段-p的核苷酸序列74:dna片段-q的核苷酸序列75:dna片段的核苷酸序列76-77:引物78:t7启动子的核苷酸序列79-90:引物序列表<110>味之素株式会社<120>生产rna的方法<130>d997-18026<150>jp2017-063777<151>2017-03-28<150>ru2017129988<151>2017-08-24<160>90<170>patentin版本3.5<210>1<211>35<212>dna<213>人工序列<220><223>引物<400>1aaaacgacggccagtggctgaatctgagcgcctgg35<210>2<211>35<212>dna<213>人工序列<220><223>引物<400>2tcaacttcaggcagtaggaacttctccaaaccagc35<210>3<211>22<212>dna<213>人工序列<220><223>引物<400>3actgcctgaagttgaggtggtg22<210>4<211>35<212>dna<213>人工序列<220><223>引物<400>4gaccatgattacgccaagaaaatcgacactgtcag35<210>5<211>26<212>dna<213>人工序列<220><223>引物<400>5ggcgtaatcatggtcatagctgtttc26<210>6<211>24<212>dna<213>人工序列<220><223>引物<400>6actggccgtcgttttacaacgtcg24<210>7<211>22<212>dna<213>人工序列<220><223>引物<400>7ggctgaatctgagcgcctggtc22<210>8<211>24<212>dna<213>人工序列<220><223>引物<400>8aagaaaatcgacactgtcagccag24<210>9<211>38<212>dna<213>人工序列<220><223>引物<400>9gacgttgatcggcacgacgatcctttttaacccatcac38<210>10<211>39<212>dna<213>人工序列<220><223>引物<400>10agacgtagacaagctcatatgggattcacctttatgttg39<210>11<211>24<212>dna<213>人工序列<220><223>引物<400>11agcttgtctacgtctgatgctttg24<210>12<211>23<212>dna<213>人工序列<220><223>引物<400>12gtgccgatcaacgtctcattttc23<210>13<211>139<212>dna<213>噬菌体bfk20<400>13gatctactcgttactcaaggcaaggtcgagcgggacggtcgaaccagcttcaagcgaccg60gatgagtatgttacagtagatagcgagcgggagaccgctcgaccttagttctcctgttgc120gggggagttcatgggatcc139<210>14<211>50<212>dna<213>噬菌体bfk20<400>14atagcataaaataacgccccaccttcttaacgggaggtggggcgttattt50<210>15<211>18<212>dna<213>人<400>15gggcattgcactccgccc18<210>16<211>259<212>dna<213>人工序列<220><223>u1ainsertrna转录单元<400>16gatctactcgttactcaaggcaaggtcgagcgggacggtcgaaccagcttcaagcgaccg60gatgagtatgttacagtagatagcgagcgggagaccgctcgaccttagttctcctgttgc120gggggagttcatgggatccacgtaccctgcgagacaggagtaatcctaaacagggcattg180cactccgcccttgctagcatagcataaaataacgccccaccttcttaacgggaggtgggg240cgttatttttacggggatc259<210>17<211>39<212>dna<213>人工序列<220><223>引物<400>17taggcaccccaggctgatctactcgttactcaaggcaag39<210>18<211>35<212>dna<213>人工序列<220><223>引物<400>18ggaagcggaagaagcgatccccgtaaaaataacgc35<210>19<211>21<212>dna<213>人工序列<220><223>引物<400>19gcttcttccgcttcctcgctc21<210>20<211>20<212>dna<213>人工序列<220><223>引物<400>20agcctggggtgcctaatgag20<210>21<211>346<212>dna<213>茄二十八星瓢虫<400>21cctcggaatcggcgaccagacgttgtgcttctactgcggcggcggtctgaaagattgggt60cgaagaagacgatccgtgggaacagcacgcgctttggttcccccagtgtaattatctatt120attgaagaaaacacccgctttcgtcaaagacgtccaagaaaaacataaaggcgatttgtc180gtcatccaagcaaaacgagaccgaagtggtagcaagtagtagcagtagtcacaactccaa240agaatctccaagtgcggtggtagaagagcgagaaagaaacaacgcagaggaaagctcgac300attatgcaaaatatgttataaaaatgaattggctgttgtatttcta346<210>22<211>36<212>dna<213>人工序列<220><223>引物<400>22gctgttgtatttctaaaggccaggaaccgtaaaaag36<210>23<211>36<212>dna<213>人工序列<220><223>引物<400>23taggcaccccaggctgatctactcgttactcaaggc36<210>24<211>23<212>dna<213>人工序列<220><223>引物<400>24tcgctatctactgtaacatactc23<210>25<211>34<212>dna<213>人工序列<220><223>引物<400>25tacagtagatagcgacctcggaatcggcgaccag34<210>26<211>21<212>dna<213>人工序列<220><223>引物<400>26tagaaatacaacagccaattc21<210>27<211>21<212>dna<213>人工序列<220><223>引物<400>27aaggccaggaaccgtaaaaag21<210>28<211>36<212>dna<213>人工序列<220><223>引物<400>28acggttcctggccttgatctactcgttactcaaggc36<210>29<211>38<212>dna<213>人工序列<220><223>引物<400>29taggcaccccaggcttcgctatctactgtaacatactc38<210>30<211>42<212>dna<213>人工序列<220><223>引物<400>30ggtaccggatcccctcgagtcgctatctactgtaacatactc42<210>31<211>30<212>dna<213>人工序列<220><223>引物<400>31ccaggtaccgatctactcgttactcaaggc30<210>32<211>31<212>dna<213>人工序列<220><223>引物<400>32cgaactcgagtagaaatacaacagccaattc31<210>33<211>25<212>dna<213>人工序列<220><223>引物<400>33gtagctcgcacgggggtttgtcttg25<210>34<211>28<212>dna<213>人工序列<220><223>引物<400>34gaactcatatgcacgggggccacataac28<210>35<211>28<212>dna<213>人工序列<220><223>引物<400>35taactcatatgcacgggggccacataac28<210>36<211>24<212>dna<213>人工序列<220><223>引物<400>36tagctcgcacgggggtttgtcttg24<210>37<211>29<212>dna<213>人工序列<220><223>引物<400>37acaactcatatgcacgggggccacataac29<210>38<211>29<212>dna<213>人工序列<220><223>引物<400>38aaaactcatatgcacgggggccacataac29<210>39<211>27<212>dna<213>人工序列<220><223>引物<400>39aactcatatgcacgggggccacataac27<210>40<211>26<212>dna<213>人工序列<220><223>引物<400>40actcatatgcacgggggccacataac26<210>41<211>28<212>dna<213>人工序列<220><223>引物<400>41aaactcatatgcacgggggccacataac28<210>42<211>20<212>dna<213>人工序列<220><223>引物<400>42ctcactggccgtcgttttac20<210>43<211>26<212>dna<213>人工序列<220><223>引物<400>43gtgagtcgtattaatttcgataagcc26<210>44<211>40<212>dna<213>人工序列<220><223>引物<400>44attaatacgactcactataggagcgagcgggagaccgctc40<210>45<211>38<212>dna<213>人工序列<220><223>引物<400>45aaggggttatgctaggctagcaagggcggagtgcaatg38<210>46<211>63<212>dna<213>人工序列<220><223>引物<400>46ctagcataaccccttggggcctctaaacgggtcttgaggggttttttgctcactggccgt60cgt63<210>47<211>63<212>dna<213>人工序列<220><223>引物<400>47acgacggccagtgagcaaaaaacccctcaagacccgtttagaggccccaaggggttatgc60tag63<210>48<211>24<212>dna<213>人工序列<220><223>引物<400>48atttcgataagccaggttgcttcc24<210>49<211>36<212>dna<213>人工序列<220><223>引物<400>49ctggcttatcgaaattaggcaccccaggcttaatac36<210>50<211>37<212>dna<213>人工序列<220><223>引物<400>50acgacggccagtgagacggttcctggcctttaatacg37<210>51<211>744<212>dna<213>谷氨酸棒杆菌<400>51gtgagcaggaaaaagaatcgcctcaccggggtagacgcactcaatgaagcattcgatgca60gtagatcatcagccgctgcttgaccaccttggtgtggacatccagcgcgatctgttggtg120cttgcgttgactcaccgctctttcgccaacgaaaacggcatgctgcccaataatgagcgc180ttggagttcctcggcgacgccgtcttgggtctctccgtggccaacaagctctatgagcag240taccccagcagccctgaatctgatgtctccaagatgcgcgcttcaattgtcagccgttac300ggcctggcagatatcgctcgcgaaattgatcttggcaaccacatattgctgggcaaaggc360gaattgctcaccgaaggtcgcagtaaggattccattcttgcggacaccacagaggcgtta420ttcggcgcgattttccgccagcacggttttgaaaccgcccgcgacgtaattttgcgcctg480tttgcctacaagatcgataacgcatcggccaggggcattcaccaggactggaagaccacg540ctgcaggaggaacttgcccagcgcaagcgccccatggctgaatattccgccacctcagtc600ggtccggatcacgatctagtgttcaccgccatcgtgacgctggaaggtgaagaaatgggt660cggggagaaggcccgaacaagaagctggccgagcaggaagcagcgcaccaggcattccga720aagcttcgggagtcccgtgcctga744<210>52<211>247<212>prt<213>谷氨酸棒杆菌<400>52metserarglyslysasnargleuthrglyvalaspalaleuasnglu151015alapheaspalavalasphisglnproleuleuasphisleuglyval202530aspileglnargaspleuleuvalleualaleuthrhisargserphe354045alaasngluasnglymetleuproasnasngluargleuglupheleu505560glyaspalavalleuglyleuservalalaasnlysleutyrglugln65707580tyrproserserprogluseraspvalserlysmetargalaserile859095valserargtyrglyleualaaspilealaarggluileaspleugly100105110asnhisileleuleuglylysglygluleuleuthrgluglyargser115120125lysaspserileleualaaspthrthrglualaleupheglyalaile130135140pheargglnhisglyphegluthralaargaspvalileleuargleu145150155160phealatyrlysileaspasnalaseralaargglyilehisglnasp165170175trplysthrthrleuglnglugluleualaglnarglysargpromet180185190alaglutyrseralathrservalglyproasphisaspleuvalphe195200205thralailevalthrleugluglygluglumetglyargglyglugly210215220proasnlyslysleualagluglnglualaalahisglnalaphearg225230235240lysleuarggluserargala245<210>53<211>405<212>dna<213>马铃薯纺锤纤块茎类病毒<400>53tctagacggaactaaactcgtggttcctgtggttcacacctgacctcctgagcagaaaag60aaaaaagaaggcggctcggaggagcgcttcagggattcccggggaaacctggagcgaact120ggcaacaaggacggtggggagtgcccagcggccgacaggagtaattcccgccgaaacagg180gttttcacccttcctttcttcgggtgtccttcctcgcgcccgcaggaccacccctcgccc240cctttgcgctgtcgcttcggctactacccggtggaaacaactgaagctcccgagaaccgc300tttttctctatcttacttgcttcggggcgagggtgtttagcccttggaaccgcagttggt360tcctggatccggggaattgttatttgtttataattttccctcgag405<210>54<211>45<212>dna<213>人工序列<220><223>引物<400>54caacatggtacctctggaacgtacatgccaccgctgagcaataac45<210>55<211>37<212>dna<213>人工序列<220><223>引物<400>55ctgcaggtcgaccaatccggatatagttcctcctttc37<210>56<211>33<212>dna<213>人工序列<220><223>引物<400>56gaatatggtaccggagctgactgggttgaaggc33<210>57<211>36<212>dna<213>人工序列<220><223>引物<400>57ctacatggtaccaatccggatatagttcctcctttc36<210>58<211>1464<212>dna<213>人工序列<220><223>dna片段<400>58gggggtagtttttattcccctagtggtttttcagtacgacaatcgagaaagacctgtttc60agccagttcgggtcatgttcgtcggtatggccacgtgcatagcgaccagttttcgagttc120actgggatttttggtgcatcgaacaagatgtaggacaatgcggtttctaggtctactttt180tgctttatgccgtacaagccccgtgggtattcagcgattgattccaaggcggcttcccag240tcctgttttgtgaaggactggcttagttctaggtctgtgtctgggtagtactgcttgttt300gtgtaagcgccgttggtgctcattgatgattcctttgaagtgtttggagttcggctagta360gtgcggcgtatggtgctgctttttgctcgtgatagctcgccttggctatgaggtcggcta420ggtaggtttccggggtgcctaggttgcgtaggtctagcaaatcccggtatgtggcctgtg480cgctgcgctggtggtgcatacagtcgttaagctgggcttttacgtctgcgatgcggtggc540ggttaggcatgttggtgtgcttcttccaagtactcacgggcgggttttgtgtatgcctgg600cgtgatgcttctttgagctgttggagttccgcttggagtgcgggtagttcgtccgcgaac660tgcttgtggtactcgtatttctcttgttcctgggcgatcagatttgcgttgaattgcagg720gcggtgagttcgtccacgcgtcgttttgctgcgttggtcatggtggcgtgccatttgcgg780ttgtggacgcggggttcaaggttgcgcacggctgcttcggctaggttggtggctgctttt840ttcagtgctcgggcttcccgttcctcgtccaacgagagcacctttggtttgttggcttcg900gctagtttttgcttctccgctttgatgagttggtcaacttcgtgttgggagaggtcgttt960ttcacgatgcgtcgaatgtggtcgttgtgggtgctgagttggtgtgagaggtagtggggt1020tctgggatttcggcgagttggtcgaggttggtgtagtgcgggttgcggcctggttggttg1080ggttcgctggggaggtcgatgtatccggttgagtctccggcgtggttgaagtgaattagg1140cgttggtagccgtattcctggttggggaggtacgacagaatgaggaagtttggtgcttct1200cctgcaatgagtcgtgcgtgttcgtagttcggtactgggtcgtgctcggggagaatgttc1260ttttgggtcatggcttctctttctgttgctctgtaagtccgtatgtgggcatgggaaagc1320cccggcaaccctttgggtcaaccggggctagatagtcgcttagaatggcttctaggctgc1380gtctcggggtgtggcaagctgtaagaggttccaactttcaccataatgaaataagatcac1440taccgggcgtattttttgagttat1464<210>59<211>161<212>dna<213>人工序列<220><223>dna片段<400>59ggtgcctatgaagctgggtatgaagatgggttaagaacagattgaaatattaacaacatg60tagtgtaatgtcttagttgtgcctttgtgagatggggccaatgggataagcgcccccggt120tgtgtagtataagaaccatgagttcctacatcaccggggcc161<210>60<211>40<212>dna<213>人工序列<220><223>引物<400>60gagttcctacatcaccggggcccgagattttcaggagcta40<210>61<211>20<212>dna<213>人工序列<220><223>引物<400>61cgccttgcgtataatatttg20<210>62<211>32<212>dna<213>人工序列<220><223>引物<400>62tctgtcgttaacggtgcctatgaagctgggta32<210>63<211>35<212>dna<213>人工序列<220><223>引物<400>63caagtaagatctatgcgacggtgtagatttcctcg35<210>64<211>37<212>dna<213>人工序列<220><223>引物<400>64ctagtaagatctcgattttcctaatcaccttgcaggt37<210>65<211>42<212>dna<213>人工序列<220><223>引物<400>65agtgtggctagcttagatctgctgtgctttcagtggatttcg42<210>66<211>41<212>dna<213>人工序列<220><223>引物<400>66tgaacagctagcaaagatctcccgatggtagtgtggggtct41<210>67<211>37<212>dna<213>人工序列<220><223>引物<400>67tatgaccatgattaccccgatggtagtgtggggtctc37<210>68<211>37<212>dna<213>人工序列<220><223>引物<400>68tctagaggatccccggctgtgctttcagtggatttcg37<210>69<211>21<212>dna<213>人工序列<220><223>引物<400>69cggggatcctctagagtcgac21<210>70<211>24<212>dna<213>人工序列<220><223>引物<400>70gtaatcatggtcatagctgtttcc24<210>71<211>20<212>dna<213>人工序列<220><223>引物<400>71agcctggggtgcctaatgag20<210>72<211>21<212>dna<213>人工序列<220><223>引物<400>72aaggccaggaaccgtaaaaag21<210>73<211>87<212>dna<213>人工序列<220><223>dna片段<400>73taggcaccccaggcttaatacgactcactataggggtaccggatcccctcgagcctatag60tgagtcgtattaaaggccaggaaccgt87<210>74<211>87<212>dna<213>人工序列<220><223>dna片段<400>74acggttcctggcctttaatacgactcactataggctcgaggggatccggtacccctatag60tgagtcgtattaagcctggggtgccta87<210>75<211>346<212>dna<213>人工序列<220><223>dna片段<400>75cctcggaatcggcgaccagacgttgtgcttctactgcggcggcggtctgaaagattgggt60cgaagaagacgatccgtgggaacagcacgcgctttggttcccccagtgtaattatctatt120attgaagaaaacacccgctttcgtcaaagacgtccaagaaaaacataaaggcgatttgtc180gtcatccaagcaaaacgagaccgaagtggtagcaagtagtagcagtagtcacaactccaa240agaatctccaagtgcggtggtagaagagcgagaaagaaacaacgcagaggaaagctcgac300attatgcaaaatatgttataaaaatgaattggctgttgtatttcta346<210>76<211>28<212>dna<213>人工序列<220><223>引物<400>76cttggtacccctcggaatcggcgaccag28<210>77<211>30<212>dna<213>人工序列<220><223>引物<400>77gttctcgagtagaaatacaacagccaattc30<210>78<211>18<212>dna<213>噬菌体t7<400>78taatacgactcactatag18<210>79<211>25<212>dna<213>人工序列<220><223>引物<400>79aaaatcgcttgaccattgcaggttg25<210>80<211>27<212>dna<213>人工序列<220><223>引物<400>80ctttagctttcctagcttgtcgttgac27<210>81<211>26<212>dna<213>人工序列<220><223>引物<400>81gcaaatcgcttgaccattgcaggttg26<210>82<211>26<212>dna<213>人工序列<220><223>引物<400>82tttagctttcctagcttgtcgttgac26<210>83<211>26<212>dna<213>人工序列<220><223>引物<400>83aaaaatcgcttgaccattgcaggttg26<210>84<211>26<212>dna<213>人工序列<220><223>引物<400>84gacaatcgcttgaccattgcaggttg26<210>85<211>26<212>dna<213>人工序列<220><223>引物<400>85ccaaatcgcttgaccattgcaggttg26<210>86<211>26<212>dna<213>人工序列<220><223>引物<400>86caaaatcgcttgaccattgcaggttg26<210>87<211>24<212>dna<213>人工序列<220><223>引物<400>87gatctactcgttactcaaggcaag24<210>88<211>28<212>dna<213>人工序列<220><223>引物<400>88gatccccgtaaaaataacgccccacctc28<210>89<211>28<212>dna<213>人工序列<220><223>引物<400>89atttttacggggatcctctagagtcgac28<210>90<211>36<212>dna<213>人工序列<220><223>引物<400>90agtaacgagtagatcgcctggggtgcctaatgagtg36当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1