通过综合组学分析研究患有转移性三阴性乳腺癌的患者的肿瘤和时间异质性的制作方法
本申请要求2017年6月1日提交的序列号为62/513,942的我们共同未决的美国临时申请的优先权。
本发明的领域是使用癌症患者的组学分析对转移性肿瘤的分子谱进行综合表征。
背景技术:
背景描述包括可用于理解本发明的信息。并不承认本文提供的任何信息是现有技术或与当前要求保护的发明相关,也不承认具体地或隐含地引用的任何出版物是现有技术。
本文中的所有出版物和专利申请都通过引用并入,其程度如同每个单独的出版物或专利申请被具体地且单独地指明通过引用并入一样。在并入的参考文献中的术语的定义或用法与本文提供的该术语的定义不一致或相反的情况下,适用本文提供的该术语的定义,而不适用该术语在该参考文献中的定义。
癌症治疗中最重大的挑战之一是对肿瘤治疗的反应存在个体差异和/或肿瘤特异性差异。尽管许多决定因素可能会影响此类个体差异或肿瘤差异,但是许多研究指出,肿瘤的固有敏感性/抗性是主要因素,这可以通过鉴定和分析肿瘤的分子谱来确定。随着全基因组测序和下一代测序平台的出现,大量数据现在可用于分子谱分析。尽管从理论角度来看当然需要大量数据,但是通常并不清楚来自大量数据的哪些类型的数据和/或哪些数据与确定对肿瘤治疗的敏感性/抗性显著相关,以及应该如何一起分析此类数据以确定关于肿瘤治疗的准确的肿瘤分子谱。
为了规避此类困难,已经努力使用选定的组学数据来确定肿瘤的分子谱。例如,ellisen的美国专利公开号2010/0035257披露,可以通过确定p63同种型和/或p73同种型的量和/或活性来鉴定对基于铂的化疗有反应的肿瘤。在另一个实例中,martin的美国专利公开号2014/0162887披露了使用多个标记基因(包括cks2、cdkn3、foxm1、rrm2等)的表达水平来确定癌症的预后。在仍另一个实例中,szallasi的美国专利公开号2016/0122827披露,可以通过确定bml和fanci基因的表达水平和/或染色体位置15q26的拷贝数来选择对肿瘤有效的疗法。然而,所有那些方法都受限于零碎信息分析,这些信息评估各个标记基因如何表达和/或染色体中特定突变的存在。因此,所有那些方法都无法使用组学数据来确定肿瘤的系统性变化,这是对肿瘤治疗的固有敏感性和/或抗性的关键属性。
因此,即使已知肿瘤中的几种常见突变和/或标记基因的表达水平与对化疗药物的敏感性和/或抗性相关,用于确定对化疗药物的敏感性和/或抗性和/或肿瘤的预后的大多数组学分析也有缺点,包括严重依赖于一些可能对肿瘤细胞的药物敏感性/抗性没有实质生理影响的标记和无法考虑许多未突变或过表达但是仍然在一些标记基因作为元素发挥作用的信号传导途径中调节药物敏感性/抗性中发挥关键作用的因子。因此,仍然需要改进的方法和系统,以使用组学数据来综合表征转移性肿瘤的分子谱。
技术实现要素:
本发明主题涉及用于使用多种类型的组学数据来综合表征转移性肿瘤的分子谱的多种方法,这些方法是通过整合多种尚未选定的与药物敏感性/抗性相关的组学数据在计算机中重构或修饰肿瘤细胞的信号传导途径来进行。因此,本发明主题的一方面包括预测在抗肿瘤治疗后患者的肿瘤预后的方法。此方法包括获得来自该患者的肿瘤样品和来自该肿瘤样品的组学数据集并且确定来自该组学数据集的基因组突变谱和多个基因的rna表达谱的步骤。优选地,该基因组突变谱包含多个体细胞突变的突变负荷。该方法继续进行获得包含多种途径元素的途径模型的步骤。然后,可以通过将该基因组突变谱和该rna表达谱整合到该途径模型中来推断经修饰的途径活性,其中至少一个与至少一种途径元素相关。经修饰的途径活性是对该抗肿瘤治疗的反应的指示,该反应可以包括但不限于对该治疗的敏感性、无反应性和获得性抗性。
最典型地,从全基因组测序、外显子组测序或rnaseq获得基因组突变谱,并且在该基因组突变谱中鉴定的该多个体细胞突变包含点突变、扩增、缺失和插入。另外,该基因组突变谱还包含具有该体细胞突变的基因组区段的拷贝数。此外,由该多个体细胞突变引起的突变负荷由在预定时间段内发生的该多个体细胞突变的数目决定。
在优选方面,用于rna表达谱分析的该多个基因中的至少两个包括rpp21、klhdc10、oxct1、nupr2、phyh、pogk、rab17、btcn1、mrpl36、marbeld2、mrps30、pla2g16、bex4、bex2、kiaa0319l、tmem25、usmg5、tyw1、dctpp1、nit2、cracr2b、st14、c1orf112、glb1l2、tomm34、nudcd3、zmym4、nupl2、lancl2、rfwd2、drosha、tmc5、znf622、zpr1、polr3a、trappc4、aifm1、ncapd3、edrf1、c11orf73、soat1、kcnk1、vps26b、cacul1、armc6、tcaim、tmem106c、polr2g、syne4、baz1b、arhgap8、tpd52l1、p2rx1、fgf2、bmp2、sntb1、abhd6。优选地,该多个基因中的至少两个参与调节干细胞的多能性的信号传导途径、癌症信号传导途径和嘧啶代谢中的至少一种。当然,应当理解,上述基因可以是野生型或突变型形式,包括错义或无义突变、插入、缺失、融合和/或易位。
关于经修饰的途径活性,设想到了经修饰的途径活性是至少一种途径元素的经修饰的蛋白质表达水平和经修饰的翻译后修饰中的至少一种。在一些实施例中,经修饰的途径活性包含myc/max途径、p53途径或ets1途径中的途径元素。优选地,使用paradigm途径分析来推断经修饰的途径活性。
另外,该方法还可以包括测量来自该肿瘤样品的蛋白质活性并且将该蛋白质活性与经修饰的途径活性进行比较的步骤。在此种方法中,该蛋白质活性包含该蛋白质的量和该蛋白质的翻译后修饰中的至少一种。在测量到的蛋白质活性不同于经修饰的途径活性或为经修饰的途径活性提供另外的信息的情况下,可以将该蛋白质活性整合到途径模型中以改善经修饰的途径活性。最后,这样修饰的途径活性可以用于产生或更新该患者的记录。
在本发明主题的又另一方面,诸位发明人设想到了一种预测抗肿瘤治疗对患者的转移性肿瘤的治疗功效的方法。在此方法中,获得来自该患者的至少两个不同的解剖位置的多个肿瘤样品和来自每个肿瘤样品的各自的组学数据集。然后,从这些组学数据集和/或从每个肿瘤样品确定来自这些组学数据集的各自的基因组突变谱和多个基因的rna表达谱。优选地,该基因组突变谱包含多个体细胞突变的突变负荷。该方法继续进行获得包含多种途径元素的途径模型的步骤。然后,可以通过将该基因组突变谱和该rna表达谱整合到该途径模型中来推断经修饰的途径活性,其中至少一个与至少一种途径元素相关。经修饰的途径活性是对该抗肿瘤治疗的反应的指示,该反应可以包括但不限于对该治疗的敏感性、无反应性和获得性抗性。然后,该方法继续使用经修饰的途径活性来产生或更新该患者的记录。该患者的记录优选地包括该抗肿瘤治疗成功治疗该转移性肿瘤的可能性和给该患者的更新治疗推荐中的至少一种。
优选地,该肿瘤是转移性三阴性乳腺癌,并且该抗肿瘤治疗是任何基于铂的疗法,例如顺铂或顺铂衍生的治疗。因此,设想到了来自至少两个不同的解剖位置的该多个肿瘤样品潜在地经由转移源自和/或衍生自相同的三阴性乳腺癌。在一些实施例中,在该抗肿瘤治疗之前和之后获得和/或在该抗肿瘤治疗期间和/或在该抗肿瘤治疗完成之后获得该多个肿瘤样品。将来自该多个肿瘤样品的获得的组学数据与匹配的正常组织(例如,该患者的健康组织等)的组学数据进行比较,以鉴定组学数据的肿瘤特异性改变。
最典型地,从全基因组测序、外显子组测序或rnaseq获得基因组突变谱,并且在该基因组突变谱中鉴定的该多个体细胞突变包含点突变、扩增、缺失和插入。另外,该基因组突变谱还包含具有该体细胞突变的基因组区段的拷贝数。此外,由该多个体细胞突变引起的突变负荷由在预定时间段内发生的该多个体细胞突变的数目决定。
在优选方面,用于rna表达谱分析的该多个基因中的至少两个包括rpp21、klhdc10、oxct1、nupr2、phyh、pogk、rab17、btcn1、mrpl36、marbeld2、mrps30、pla2g16、bex4、bex2、kiaa0319l、tmem25、usmg5、tyw1、dctpp1、nit2、cracr2b、st14、c1orf112、glb1l2、tomm34、nudcd3、zmym4、nupl2、lancl2、rfwd2、drosha、tmc5、znf622、zpr1、polr3a、trappc4、aifm1、ncapd3、edrf1、c11orf73、soat1、kcnk1、vps26b、cacul1、armc6、tcaim、tmem106c、polr2g、syne4、baz1b、arhgap8、tpd52l1、p2rx1、fgf2、bmp2、sntb1、abhd6。优选地,该多个基因中的至少两个参与调节干细胞的多能性的信号传导途径、癌症信号传导途径和嘧啶代谢中的至少一种。当然,应当理解,上述基因可以是野生型或突变型形式,包括错义或无义突变、插入、缺失、融合和/或易位。
关于经修饰的途径活性,设想到了经修饰的途径活性是至少一种途径元素的经修饰的蛋白质表达水平和经修饰的翻译后修饰中的至少一种。在一些实施例中,经修饰的途径活性包含myc/max途径、p53途径或ets1途径中的途径元素。优选地,使用paradigm途径分析来推断经修饰的途径活性。
另外,该方法还可以包括测量来自该肿瘤样品的蛋白质活性并且将该蛋白质活性与经修饰的途径活性进行比较的步骤。在此种方法中,该蛋白质活性包含该蛋白质的量和该蛋白质的翻译后修饰中的至少一种。在测量到的蛋白质活性不同于经修饰的途径活性或为经修饰的途径活性提供另外的信息的情况下,可以将该蛋白质活性整合到途径模型中以改善经修饰的途径活性。
本发明主题的仍另一方面包括一种预测患有三阴性乳腺癌的患者对顺铂治疗的反应的方法。此方法包括获得来自该患者的肿瘤样品和来自该肿瘤样品的组学数据集并且确定多个基因的rna表达谱的步骤。最优选地,该多个基因中的至少一个与调节干细胞的多能性的信号传导途径、癌症信号传导途径和嘧啶代谢中的至少一种相关。该方法继续进行获得包含多种途径元素的途径模型的步骤。然后,可以通过将该rna表达谱整合到该途径模型中来推断经修饰的途径活性,其中至少一个与至少一种途径元素相关。经修饰的途径活性是对该抗肿瘤治疗的反应的指示,该反应可以包括但不限于对该治疗的敏感性、无反应性和获得性抗性。
在优选方面,用于rna表达谱分析的该多个基因中的至少两个包括rpp21、klhdc10、oxct1、nupr2、phyh、pogk、rab17、btcn1、mrpl36、marbeld2、mrps30、pla2g16、bex4、bex2、kiaa0319l、tmem25、usmg5、tyw1、dctpp1、nit2、cracr2b、st14、c1orf112、glb1l2、tomm34、nudcd3、zmym4、nupl2、lancl2、rfwd2、drosha、tmc5、znf622、zpr1、polr3a、trappc4、aifm1、ncapd3、edrf1、c11orf73、soat1、kcnk1、vps26b、cacul1、armc6、tcaim、tmem106c、polr2g、syne4、baz1b、arhgap8、tpd52l1、p2rx1、fgf2、bmp2、sntb1、abhd6。
任选地,除了该rna表达谱外,还可以从该组学数据集确定基因组突变谱,其中该基因组突变谱包含多个体细胞突变的突变负荷,并且可以通过将该基因组突变谱和该rna表达谱整合到该途径模型中来推断经修饰的途径活性。此外,可以测量来自该肿瘤样品的蛋白质活性,其中该蛋白质活性包含该蛋白质的量和该蛋白质的翻译后修饰中的至少一种,并且可以将该蛋白质活性与经修饰的途径活性进行比较。可以将这样测量的蛋白质活性整合以改善经修饰的途径活性。
本发明主题的多个对象、特征、方面和优点将根据以下关于优选实施例的详细描述和附图而变得更清楚。
附图说明
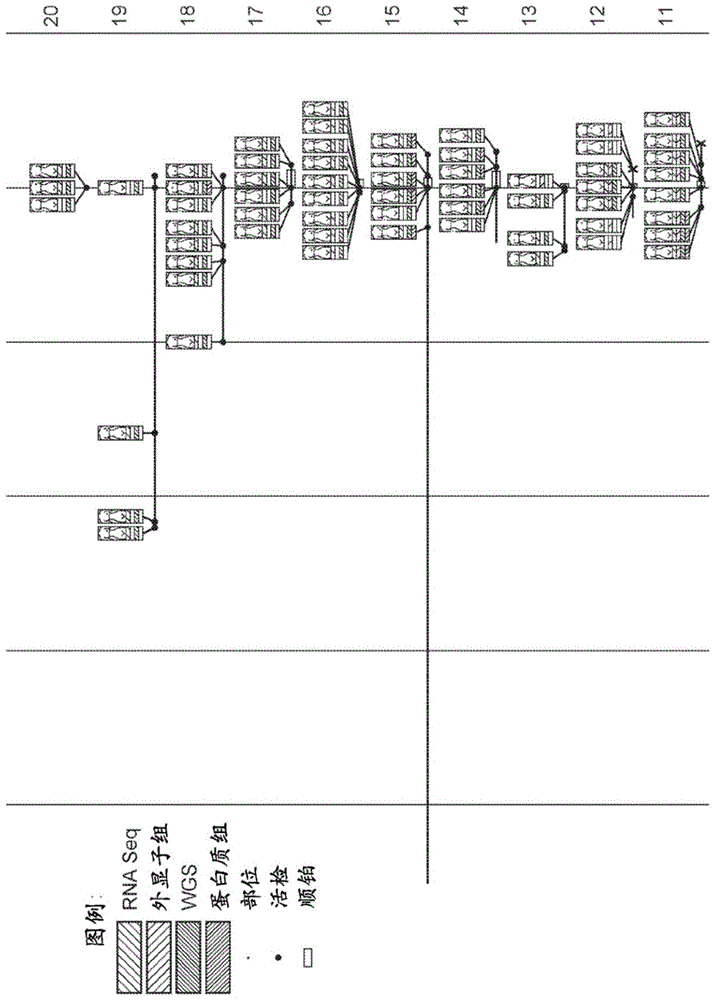
图1描绘了从中获得活检的肿瘤组织用于组学分析的20位三阴性乳腺癌(tnbc)患者的时间轴。
图2a示出了描绘在17位患者的各自活检中高可信度的基因组拷贝数畸变(cna)的条形图。
图2b示出了描绘在17位患者的各自活检中鉴定的外显子组单核苷酸变体(snv)和碱基的插入和缺失(indel)的条形图。
图2c示出了显示4位患者的顺铂前和顺铂后肿瘤组织中存在的和获得的snv的计数的文氏图。
图3a示出了在顺铂敏感患者和抗性患者之间显著差异表达的57个基因的热图。
图3b示出了提供针对癌症信号传导和嘧啶代谢途径富集的图3a的热图中所示的57个基因的表格。
图4a描述了使用paradigm途径分析产生推断的途径活性的示意图。
图4b示出了从定量质谱法观察到的蛋白质量与从10种蛋白质的paradigm推断的蛋白质活性的比较,这10种蛋白质将未经顺铂治疗的
图5a描绘了在来自4位患者的6种不同的活检中在顺铂治疗之前和之后的细胞途径活性的变化。
图5b描绘了多种细胞途径与ets1途径的关系,并且描绘了在顺铂治疗之前和之后活性发生变化的途径元素。
具体实施方式
诸位发明人设想到了在肿瘤细胞中单独地或组合地观察到的基因组、转录组和/或蛋白质组变化这样改变肿瘤细胞的细胞信号传导途径,使得肿瘤细胞显示出独特的固有生理特征,例如对药物的敏感性或抗性、对坏死或细胞凋亡的易感性、去分化和/或干细胞样特征的获得或转移性特征的获得。尽管对dna、转录物表达水平或蛋白质活性的个体变化的鉴定可能与肿瘤细胞的一些生理变化相关,并且可以用作预测细胞状态的标记,但是这种方法通常无法考虑细胞信号传导网络中此类变化或整体或净变化的个体变化,这可能导致肿瘤细胞具有相似或不同的生理特征。例如,肿瘤a和肿瘤b的基因c、d和e的表达水平可能不同(例如,肿瘤a的c和d的表达增加,而肿瘤b仅e的表达增加),然而肿瘤a和b对化疗剂f具有基本上相似的药物敏感性。在另一个实例中,肿瘤g和h的标记基因i和j的表达水平可能相似,但是由于未被鉴定为标记基因或蛋白质的其他因子,对化疗剂k具有不同的药物敏感性。
从不同的角度来看,诸位发明人发现,从患者肿瘤组织获得的多种组学数据可以共同用于确定可能影响肿瘤组织的固有特性的细胞信号传导网络的整体或净变化。另外,可以选择一组组学数据作为肿瘤细胞特定生理特征(例如,对治疗a的敏感性等)的相关数据集,使得可以高效且有效地以及时方式执行使用组学数据集的细胞信号传导网络分析。
因此,在本发明主题的一个尤其优选的方面,诸位发明人设想到了一种使用从肿瘤样品的组学数据集获得的基因组突变谱和rna表达谱预测在抗肿瘤治疗后患者的肿瘤预后的方法。由于基因组突变谱和rna表达谱与匹配的正常谱的差异,将基因组突变谱和rna表达谱整合到途径模型中以推断经修饰的途径活性。这样推断的经修饰的途径活性可以是对抗肿瘤治疗的反应的指示,并且可以进一步用于预测在抗肿瘤治疗后患者的肿瘤预后。另外,蛋白质组学数据也可以用于此类方法。
如本文所用,术语“肿瘤”是指以下项并且可与以下项互换使用:一种或多种癌细胞、癌组织、恶性肿瘤细胞或恶性肿瘤组织,它们可以位于或者发现于人体的一或多个解剖位置中。应当注意,如本文所用的术语“患者”包括经诊断患有病症(例如,癌症)的个体以及出于检测或鉴定病症的目的经历检查和/或测试的个体二者。因此,患有肿瘤的患者是指经诊断患有癌症的个体以及被怀疑患有癌症的个体二者。如本文所用,术语“提供(provide)”或“提供(providing)”是指并且包括制造、产生、放置、使得能使用、转移或使得即可使用的任何行为。
获得组学数据
设想到了获得来自患者的肿瘤样品(肿瘤细胞或肿瘤组织)(或来自患者或健康个体的健康组织作为比较)的任何合适的方法。最典型地,可以经由活检(包括液体活检、或在手术或独立的活检程序期间经由组织切除术获得的等)获得来自患者的肿瘤样品,其可以是新鲜的或经处理的(例如,冷冻的等),直到用于从组织获取组学数据的进一步过程为止。例如,肿瘤细胞或肿瘤组织可以是新鲜的或冷冻的。又例如,肿瘤细胞或肿瘤组织可以呈细胞/组织提取物的形式。在一些实施例中,肿瘤样品可以从单个或多个不同的组织或解剖区域获得。例如,可以从患者的乳房以及其他器官(例如,肝、脑、淋巴结、血液、肺等)获得转移性乳腺癌组织用作转移的乳腺癌组织。优选地,可以获得患者的健康组织或匹配的正常组织(例如,患者的非癌性乳腺组织),或者也可以经由类似的方式获得来自健康的个体(患者以外)的健康组织作为比较。
在一些实施例中,为了确定在相关时间段内肿瘤样品的任何变化,可以在多个时间点从患者获得肿瘤样品。例如,可以在样品被确定或诊断为癌性之前和之后获得肿瘤样品(或疑似肿瘤样品)。在另一个实例中,可以在一次或一系列抗肿瘤治疗(例如,放疗、化疗、免疫疗法等)之前、期间和/或之后(例如,完成后等)获得肿瘤样品(或疑似肿瘤样品)。在仍另一个实例中,在鉴定新的转移的组织或细胞后,可以在肿瘤进展期间获得肿瘤样品(或疑似肿瘤样品)。
在图1中描述了获得来自20位三阴性乳腺癌(tnbc)患者的肿瘤样品和/或来自肿瘤样品的组学数据的示例性时间轴。“组学在癌症中的强化试验”-001(“intensivetrialofomicsincancer”-001,itomic-001;美国临床试验数据库(clinicaltrials.gov)id:nct01957514)招募了未经铂治疗并且计划接受顺铂的患有转移性三阴性乳腺癌(tnbc)的患者。在完成顺铂治疗之前和之后以及任何后续疗法之后,在仔细控制的条件下进行多次活检(最多7个转移部位)。方框表示活检部位(在患者体内的位置)、时间安排、顺铂治疗以及所获得的组学数据类型。研究日期0是纳入试验的第一个日期。图1所示的大多数患者都有先前的乳腺癌诊断,因此在第0天之前有时间轴。许多患者有多个历史活检,将其作为试验的一部分进行了分析。如图所示,在不同的时间点在身体的不同位置(例如,左或右乳房、卵巢、小腹等)进行肿瘤组织的活检。从那些活检的样品中的一些中,通常基于活检类型、组织可利用性、组织状态、给定时间段所需的相关信息等获得不同类型或组合类型的组学数据。
从获得的肿瘤细胞或肿瘤组织中,可以将dna(例如,基因组dna、染色体外dna等)、rna(例如,mrna、mirna、sirna、shrna等)和/或蛋白质(例如,膜蛋白、胞质蛋白、核蛋白等)分离并且进一步分析以获得组学数据。可替代地和/或另外地,获得组学数据的步骤可以包括从存储一位或多位患者和/或健康个体的组学信息的数据库接收组学数据。例如,可以从患者肿瘤组织中分离的dna、rna和/或蛋白质获得患者肿瘤的组学数据,并且所获得的组学数据可以与患有相同类型的肿瘤或不同类型的肿瘤的其他患者的其他组学数据集一起存储在数据库(例如,云数据库、服务器等)中。从健康个体或患者的匹配的正常组织(或健康组织)获得的组学数据也可以存储在数据库中,使得在分析时可以从数据库中检索相关的数据集。同样,在获得蛋白质数据的情况下,这些数据也可以包括蛋白质活性,特别是在蛋白质具有酶活性(例如,聚合酶、激酶、水解酶、裂解酶、连接酶、氧化还原酶等)的情况下。
如本文所用,组学数据包括但不限于与基因组学、蛋白质组学和转录组学以及特定的基因表达或转录物分析和细胞的其他特征和生物学功能相关的信息。关于基因组学数据,合适的基因组学数据包括dna序列分析信息,其可以通过肿瘤和匹配的正常样品的全基因组测序和/或外显子组测序(典型地在至少10倍、更典型地至少20倍的覆盖深度下)获得。可替代地,还可以从来自先前序列测定的已建立的序列记录(例如,sam、bam、fasta、fastq或vcf文件)提供dna数据。因此,数据集可以包括未处理的或已处理的数据集,并且示例性数据集包括具有bam格式、sam格式、fastq格式或fasta格式的那些。然而,尤其优选的是,以bam格式或作为bambamdiff对象提供数据集(例如,us2012/0059670a1和us2012/0066001a1)。组学数据可以源自全基因组测序、外显子组测序、转录组测序(例如,rna-seq)或源自基因特异性分析(例如,pcr、qpcr、杂交、lcr等)。同样,可以按多种方式进行序列数据的计算分析。然而,在最优选的方法中,如例如在us2012/0059670a1和us2012/0066001a1中披露的使用bam文件和bam服务器通过对肿瘤和正常样品进行位置引导的同步比对在计算机中进行分析。这样的分析有利地减少假阳性新表位,并且显著地减少对存储器和计算资源的需求。
关于患者的肿瘤和匹配的正常组织的分析,认为许多方式适用于本文,只要此类方法将能够在肿瘤和匹配的正常序列之间产生差异序列对象或位置特异性差异的其他识别即可。示例性方法包括与外部参考序列(例如,hg18或hg19)的序列比较、与内部参考序列(例如,匹配的正常序列)的序列比较以及与已知的常见突变模式(例如,snv)的序列处理。因此,在肿瘤和匹配的正常、肿瘤和液体活检以及匹配的正常和液体活检之间检测突变的设想到的方法和程序包括icallsv(url:github.com/rhshah/icallsv)、varscan(url:varscan.sourceforge.net)、mutect(url:github.com/broadinstitute/mutect)、strelka(url:github.com/illumina/strelka)、somaticsniper(url:gmt.genome.wustl.edu/somatic-sniper/)和bambam(us2012/0059670)。
然而,在本发明主题的尤其优选的方面,通过第一序列数据(肿瘤样品)与第二序列数据(匹配的正常样品)的增量同步比对来进行序列分析,例如使用如例如描述于cancerres[癌症研究]2013年10月1日;73(19):6036-45、us2012/0059670和us2012/0066001的算法以如此产生患者和肿瘤的特定突变数据。如将容易理解的,序列分析也可以按这样的方法进行,将来自肿瘤样品的组学数据与匹配的正常组学数据进行比较,以便如此进行如下分析,其不仅可以告知使用者对于患者体内的肿瘤而言真正的突变,还可以告知使用者在治疗期间新出现的突变(例如,经由匹配的正常和匹配的正常/肿瘤的比较、或经由肿瘤的比较)。另外,使用此类算法(尤其是bambam),可以容易地确定特定突变的等位基因频率和/或克隆群体,这可以有利地提供关于特定肿瘤细胞部分或群体的治疗成功的指示。因此,组学数据分析可以揭示错义和无义突变、拷贝数变化、杂合性丢失、缺失、插入、倒位、易位、微卫星变化等。
此外,应当注意,数据集优选地反映了同一患者的肿瘤和匹配的正常样品,以便如此获得患者和肿瘤的具体信息。因此,可以排除不引起肿瘤的遗传种系改变(例如,沉默突变、snp等)。当然,应当认识到,肿瘤样品可能来自初始肿瘤、来自开始治疗时的肿瘤、来自复发性肿瘤或转移部位等。在大多数情况下,患者的匹配的正常样品可能是血液或来自与肿瘤相同的组织类型的未患病的组织。
另外,癌症和/或正常细胞的组学数据包含如下转录组数据集,其包括从患者(最优选地从患者的癌症组织(患病组织)和匹配的健康组织)或健康个体获得的一种或多种rna(优选细胞mrna)的序列信息和表达水平(包括表达分析或剪接变体分析)。本领域已知许多转录组分析方法,并且所有已知方法都被认为适用于本文(例如,rnaseq、rna杂交阵列、qpcr等)。因此,优选的材料包括mrna和初级转录物(hnrna),并且rna序列信息可以从逆转录的polya+-rna获得,该逆转录的polya+-rna进而从同一患者的肿瘤样品和匹配的正常(健康的)样品获得。同样,应当注意,尽管典型地优选polya+-rna作为转录组的代表,但是其他形式的rna(hn-rna、非多聚腺苷酸化rna、sirna、mirna等)也被认为适用于本文。优选的方法包括定量rna(hnrna或mrna)分析和/或定量蛋白质组学分析,尤其是包括rnaseq。在其他方面,使用基于rna-seq、qpcr和/或rtpcr的方法进行rna定量和测序,尽管多种可替代的方法(例如,基于固相杂交的方法)也被认为是合适的。从另一个角度来看,转录组学分析(单独地或与基因组分析组合)可能适合于鉴定和定量具有癌症特异性突变和患者特异性突变的基因。
应当理解,可以针对特定疾病、疾病阶段、特定突变或甚至基于个人突变谱或所表达的新表位的存在来选择一种或多种所需核酸。可替代地,在需要发现或扫描新突变或特定基因的表达变化的情况下,可以用rnaseq替代实时定量pcr,以便如此覆盖患者转录组的至少一部分。此外,应当理解,可以静态地进行分析,或者在重复取样的时间过程中进行分析以获得动态图片,而无需对肿瘤或转移进行活检。
此外,癌症和/或正常细胞的组学数据包含如下蛋白质组学数据集,其包括蛋白质表达水平(蛋白质分子的定量)、翻译后修饰、蛋白质-蛋白质相互作用、蛋白质-核苷酸相互作用、蛋白质-脂质相互作用等。因此,还应当理解,如本文呈现的蛋白质组学分析还可以包括选定的蛋白质的活性测定。可以从新鲜切除的组织、从冷冻或以其他方式保存的组织、甚至从ffpe组织样品进行这样的蛋白质组学分析。最优选地,蛋白质组学分析是定量的(即,提供表达的多肽的定量信息)和定性的(即,提供多肽的数字或定性指定活性)。设想到了任何合适类型的分析。然而,特别优选的蛋白质组学方法包括基于抗体的方法和质谱方法。此外,应当注意,蛋白质组学分析不仅可以提供有关蛋白质本身的定性或定量信息,而且也可以包括蛋白质具有催化活性或其他功能活性的蛋白质活性数据。用于进行蛋白质组学测定的一种示例性技术描述于通过引用并入本文的us7473532中。鉴定并且甚至定量蛋白质表达的进一步合适的方法包括多种质谱分析(例如,选择反应监测(srm)、多反应监测(mrm)和连续反应监测(crm))。
组学数据分析
诸位发明人设想到了可以使用组学数据、优选地两种或更多种类型的组学数据来确定肿瘤组织的分子谱或分子标签。尽管可以使用任何类型或亚型的组学数据来确定肿瘤组织的分子谱或分子标签,但是设想到了优选的组学数据的类型可以基于肿瘤的类型、基于所需信息(例如,关于固有药物敏感性、肿瘤细胞干细胞特性等的信息)和/或肿瘤的预后(例如,转移的、免疫抗性的等)而变化。可能与肿瘤发展相关的基因组学数据的示例性亚型可以包括但不限于基因组扩增(如代表的基因组拷贝数畸变)、体细胞突变(例如,点突变(例如,无义突变、错义突变等)、缺失、插入等)、基因组重排(例如,染色体内重排、染色体外重排、易位等)、染色体外基因组的出现和拷贝数(例如,双微染色体等)。另外,基因组数据还可以包括肿瘤突变负荷,其通过在预定时间段内或相关时间段内由肿瘤细胞携带的或出现在肿瘤细胞中的突变的数目来测量。
例如,图2a-2c示出了从未接受基于铂的治疗和在试验期间接受顺铂的17位转移性三阴性乳腺癌患者获得的66个活检中鉴定出的肿瘤突变负荷。图2a展示了在每个个体活检中与高度可信的基因组拷贝数畸变(cna)重叠的基因的数目、以及在顺铂治疗之前和之后每位患者的具有cna的基因的总数。基因组扩增定义为中位拷贝数估计值高于6个拷贝的>5kb的基因组区域,而基因组缺失定义为中位拷贝数估计值低于1个拷贝。在上方的条形图中绘制了基因组中检测到的扩增事件,并且在下方的条形图中绘制了基因组中检测到的缺失事件。拷贝数估计值源自如通过循环二元分割(cbs)和肿瘤纯度/倍性估计值报告的相对覆盖。诸位发明人观察到,一些个体转移瘤经历了相对较多的cna事件。略有阴影(从左上到右下)的是在顺铂治疗之前,并且有浓密阴影(从左下到右上)的是在顺铂治疗之后。
图2b展示了从17位转移性三阴性乳腺癌患者的每个活检中看到的可靠的外显子组单核苷酸变体(snv)的数目(上)和碱基的插入和缺失(indel)的数目(下)、以及每位患者的snv和indel的总数。如上所述,通过比较从肿瘤细胞和匹配的正常组织获得的外显子组序列数据来检测snv和indel。通过应用几个过滤步骤来进一步处理这样鉴定出的突变,以产生突变(存在于基因组或外显子组中而不仅仅是测序错误的真正的体细胞突变、种系突变或等位基因变异等)的可靠调用。因此,示例性过滤可以基于读取支持物的数目、碱基质量、可以从单核苷酸多态性数据库(dbsnp)鉴定出的单核苷酸多态性(snp)以及体细胞与种系变体调用。诸位发明人发现,在此项研究中的tnbc患者倾向于在以后的活检中缓慢获得snv和indel计数。然而,患者之间的顺铂治疗后初始突变负荷显著不同(突变负荷幅度高达10倍),这表明肿瘤突变负荷可能是患者特异性因素,从而响应于顺铂治疗影响肿瘤组织或肿瘤细胞的固有特性。
为了证实顺铂治疗对肿瘤突变负荷的作用,在顺铂疗法之前和治疗之后立即从4位患者获得活检的肿瘤组织。从每个活检的组织获得全基因组测序数据,并且分析顺铂治疗之前和之后获得的snv数目。图2c描绘了在顺铂前和顺铂后活检中存在的和获得的snv的计数。在顺铂治疗后出现的snv可能指示响应于顺铂治疗的肿瘤进化(例如,获得的敏感性或抗性等)。在顺铂治疗后,显著干扰了超过300个基因的表达,并且每位患者平均引入了270个突变。
除了基因组学数据外,一种或多种亚型的转录组学数据也可以用于确定肿瘤组织的分子谱或分子标签。示例性转录组学数据包括但不限于如通过mrna的量、mrna的成熟水平(例如,polya尾的存在等)和/或转录物的剪接变体测量的多个mrna的表达水平。基因的数目(至少两个、至少五个、至少十个、至少十五个等)、转录物或rna的类型(mrna、mirna等)或用于确定肿瘤组织的分子谱或分子标签的基因的选择可以基于肿瘤的类型、基于所需的信息(例如,关于固有药物敏感性、肿瘤细胞干细胞特性等的信息)和/或肿瘤的预后(例如,转移的、免疫抗性的等)而变化。例如,用于确定与肿瘤干细胞特性相关的分子标签的基因选择和/或基因数目可以不同,或和用于确定与细胞对特定化疗药物的敏感性相关的分子标签的基因选择和/或基因数目具有最小重叠。设想到了要包括在相关转录组学数据集中以区分肿瘤样品(从匹配的正常样品中或在具有不同生理特征的肿瘤样品中)的基因可以包括任何肿瘤特异性基因、炎症相关基因、dna修复相关基因(例如,碱基切除修复、错配修复、核苷酸切除修复、同源重组、非同源末端连接等)、与对dna损伤剂的敏感性相关的基因、dna复制机器相关基因。然而,还设想到了要包括在相关转录组学数据集中以区分肿瘤样品的基因可以包括与疾病无关的基因(例如,管家基因),包括但不限于与转录因子、rna剪接、trna合成酶、rna结合蛋白、核糖体蛋白或线粒体蛋白或者非编码rna(例如,微小rna、小干扰rna、长非编码rna(lncrna)等)相关的那些基因。
图3a提供了57个基因的示例性rna表达分析(显示为热图),这57个基因在顺铂敏感肿瘤细胞中与顺铂抗性肿瘤细胞相比差异表达(使用benjamini-hochberg调整q<0.05,调整后的p值p<0.05)。在此实例中,从14位患者(8位顺铂敏感患者和6位顺铂抗性患者)获得了总共32个活检的肿瘤组织,并且比较了这32个活检的肿瘤组织的转录组学数据,以鉴定在顺铂敏感肿瘤细胞和顺铂抗性肿瘤细胞中差异表达的基因。定量表达水平并且表示为z得分(z=(肿瘤样品中的表达-参考样品中的平均表达)/参考样品中表达的标准差)。诸位发明人发现,衍生自基因rpp21、klhdc10、oxct1、nupr2、phyh、pogk、rab17、btcn1、mrpl36、marbeld2、mrps30、pla2g16、bex4、bex2、kiaa0319l、tmem25、usmg5、tyw1、dctpp1、nit2、cracr2b、st14、c1orf112、glb1l2、tomm34、nudcd3、zmym4、nupl2、lancl2、rfwd2、drosha、tmc5、znf622、zpr1、polr3a、trappc4、aifm1、ncapd3、edrf1、c11orf73、soat1、kcnk1、vps26b、cacul1、armc6、tcaim、tmem106c、polr2g、syne4、baz1b、arhgap8、tpd52l1、p2rx1、fgf2、bmp2、sntb1和abhd6的转录物的表达水平在顺铂敏感和顺铂抗性肿瘤样品中显著不同。在这57个基因中,衍生自rpp21、klhdc10、oxct1、nupr2、phyh、pogk、rab17、btcn1、mrpl36、marbeld2、mrps30、pla2g16、bex4、bex2、kiaa0319l、tmem25、usmg5、tyw1、dctpp1、nit2、cracr2b、st14、c1orf112、glb1l2、tomm34、nudcd3、zmym4、nupl2、lancl2、rfwd2、drosha、tmc5、znf622、zpr1、polr3a、trappc4、aifm1、ncapd3、edrf1、c11orf73、soat1、kcnk1、vps26b、cacul1、armc6、tcaim、tmem106c、polr2g、syne4、baz1b、arhgap8和tpd52l1的转录物在顺铂敏感肿瘤样品中的表达水平更高,并且与顺铂抗性肿瘤样品相比,衍生自p2rx1、fgf2、bmp2、sntb1和abhd6的转录物在顺铂敏感肿瘤样品中的表达水平更低。因此,诸位发明人设想到了用于确定转移性tnbc细胞关于顺铂敏感性或抗性的分子谱或标签的rna表达谱可以包括以上鉴定出的57个基因中的至少5个基因、至少10个基因、至少15个基因、至少20个基因或至少有25个基因。
任选地,一种或多种亚型的蛋白质组学数据可以用于确定肿瘤组织的分子谱或分子标签。示例性蛋白质组学数据包括但不限于一种或多种蛋白质或肽的量、一种或多种蛋白质或肽的翻译后修饰(例如,磷酸化、糖基化、形成二聚体、泛素化等)和/或蛋白质或肽的亚细胞定位。例如,图4b示出了10种蛋白质(aldha1、kras2b、sparc、p16、pten、rrm1、ido1、egfr、ercc1、her2)的一种示例性蛋白质组学分析,这10种蛋白质的表达水平将未经顺铂治疗的肿瘤样品与经顺铂治疗的样品显著地区分开(通过fisher精确检验测得)。因此,诸位发明人设想到了用于确定转移性tnbc细胞关于顺铂敏感性或抗性的分子谱或标签的蛋白质组学分析可以包括以上列示的至少一种、至少两种、至少五种或至少七种蛋白质。可替代地并且更优选地,诸位发明人设想到了肿瘤细胞的蛋白质组学分析可以用作确证数据以改善以下描述的途径分析。例如,可以从数据集中滤除或去除与测量到的蛋白质组学数据不匹配的推断的途径活性(例如,推断的途径活性指示在顺铂治疗后是有活性的或量增加,而测量到的蛋白质组学数据显示在顺铂治疗后是受抑制的或量减少等)。
途径分析
不希望受任何特定理论的束缚,诸位发明人设想到了肿瘤组织的突变谱和/或rna表达谱独立地或共同地影响细胞内信号传导网络,从而可能改变肿瘤组织或细胞的固有特性。因此,在本发明主题的一个优选方面,可以将如此确定的肿瘤组织的突变谱和/或rna表达谱整合到途径模型中,以产生经修饰的途径或肿瘤特异性途径。最典型地,该途径模型包含通过一个或多个调节节点连接的多种途径元素(例如,蛋白质)。例如,途径模型[a]是基于因子图的途径模型(例如,paradigm途径模型),其包含通过调节节点i(在元素a和b之间)以及另一个调节节点ii(在元素b和c之间)连接的途径元素a、b和c(a-i-b-ii-c)。调节节点i和ii代表可能会影响b和c的活性的除a或b以外的任何因子。因此,途径模型[a]可以经由调节节点i和ii之一与另一个途径模型[b]耦合。因此,在一些实施例中,该途径模型可以包括单个途径(例如,pka介导的细胞凋亡途径等)。因此,在一些实施例中,该途径模型可以是单度模型,其包括一个或多个彼此平行或基本独立的信号传导途径。在其他实施例中,该途径模型可以是多度模型,其可以包括经由一个或多个调节节点耦合的多个信号传导途径(例如,具有途径[a]和[b]的二度模型,其中途径[a]和[b]在途径[a]的调控节点耦合;具有途径[a]、[b]和[c]的三度模型,其中途径[a]和[b]在途径[a]的调控节点耦合,并且途径[b]和[c]在途径[b]的调节节点耦合)。
可以如在wo2014/193982中所述的在中心法则模块中使用组学数据作为输入来推断或计算每种途径元素的途径元素活性(dna-rna-蛋白质-蛋白质活性),将该专利通过引用并入本文。例如,在编码蛋白质a的基因在外显子组中携带多个基因组突变,并且在药物治疗后该基因的rna表达水平增加的情况下,可以从此类基因组和转录组学谱推断出,由于关键的翻译后修饰残基中的错义突变,该蛋白质的量可能会增加,而此种蛋白质的活性可能在信号传导途径(其中蛋白质a是该信号传导途径的元素)中提供显性负作用。基于这样推断的个体途径元素活性,可以在同一信号传导途径或通过调节节点连接的另一个信号传导途径中推断下游信号传导途径元素的活性。
因此,可以将各种类型的组学数据整合到单个途径模型中,以便如此允许在经测量的属性(例如,dna拷贝数和/或突变、rna转录水平、蛋白质量和/或活性)的基础上计算推断的属性(例如,没有从样品获得数据的dna拷贝数和/或突变、rna转录水平、蛋白质量和/或活性),并且还计算推断的途径活性。有利地,此类计算可以利用全部可用的组学数据,或者仅使用与相应的正常值有显著偏差的组学数据(例如,由于拷贝数变化、过表达或低表达、蛋白活性丧失等)。使用此种系统,应当理解,可以检测细胞信号传导活性和此类信号传导途径的变化而不是仅分析单个或多个标记,否则当仅考虑单个或多个标记而无视其功能时,这些信号传导活性和这些信号传导途径的变化将不会被注意。
优选地,可以经由机器学习算法(例如,线性核svm、一阶多项式核svm、二阶多项式核svm、岭回归、lasso、弹性网络、序贯最小优化、随机森林法、j48树、朴素贝叶斯、jrip规则、hyperpipes和nmfpredictor)用健康个体的组学数据作为输入和确证数据来预训练途径模型。在这样的实施例中,通过机器学习算法,将用权重和方向提供每种途径元素和调节节点的因子,以确定下游途径元素的活性。例如,在途径元素a和b连接至调节节点i的情况下,整合或计算途径元素a的量(例如,拷贝数、rna的表达水平)和/或状态(例如,突变的类型和位置、磷酸化蛋白质的磷酸化数目等)和/或调节节点i的任何因子(例如,影响途径元素a活性的酶的活性等)中的每个或至少一个,以推断途径元素b的活性(例如,蛋白质b的量、状态)。
因此,这样训练的途径模型可以用作预测肿瘤组织中途径或途径元素将如何变化的模板。如图4a所描绘,可以使用paradigm将从患者获得的组学数据(并且优选地与匹配的正常组织或来自健康个体的健康组织比较)整合到基于因子图的模型中,以推断或预测与匹配的正常组织或来自健康个体的健康组织相比,由于肿瘤特异性组学数据的变化,哪些途径元素将变化以及将如何变化。尽管图4a将paradigm描绘为使用组学数据的示例性途径分析方法和/或工具,但是应当注意,可以这样进行机器训练并且产生可靠输出数据的任何合适的途径模型都被认为是适当的。因此,合适的途径模型包括基于基因集合富集分析(gsea,布罗德研究所(broadinstitute))的模型、基于信号传导途径影响分析(spia,bioconductor)的模型和病理学家途径模型(ncbi)以及基于因子图的模型,尤其是如在均通过引用并入本文的wo2011/139345a2、wo2013/062505a1和wo2014/059036中描述的paradigm。
可能与肿瘤预后相关或密切相关的信号传导途径的数目和类型和/或细胞网络的范围可能会根据肿瘤的类型、肿瘤的分期和/或分析目的(例如,药物敏感性、肿瘤细胞干细胞特性、免疫抗性等)而变化。换言之,将要使用组学数据进行分析的信号传导途径的数目和类型可能限于被预先确定为与组学数据的变化相关的那些,或被鉴定为与组学数据的变化最相关的或最受影响的那些。因此,在一个实施例中,可以基于信号传导途径元素的数目和/或对信号传导途径中信号传导途径元素的影响程度来选择信号传导途径,该影响程度由肿瘤样品中基因的差异表达表示。例如,诸位发明人发现,如图3b所示,在顺铂敏感和抗性肿瘤之间显示出差异表达水平的许多基因是几种信号传导途径(例如,调节干细胞的多能性的信号传导途径、癌症途径、rna聚合酶调节途径、嘧啶代谢途径)的途径元素,这表明那些信号传导途径可能会在作为信号传导途径元素的基因的突变和/或表达谱发生变化时而被修饰。
尽管信号传导途径元素不存在于那些信号传导途径中,但是一些信号传导途径可能受该信号传导途径元素的改变的实质影响。因此,可替代地和/或另外地,由于组学数据的变化,可以基于对途径的总体影响来选择信号传导途径的数目和类型。在此类实施例中,可以使用机器学习算法和一个或多个可变因子(例如,如图3a所示的差异表达基因)来检查可能存在于肿瘤细胞中的多个信号传导途径,并且可以选择最受组学数据的变化的影响的信号传导途径。
tnbc患者肿瘤的中心法则模块中的途径元素活性(dna-rna-蛋白质-蛋白质活性)的示例性预测在图4b中示出,该图描绘了使用定量质谱法测量的来自肿瘤样品的蛋白质量(o)以及从前10种蛋白质(aldha1、kras2b、sparc、p16、pten、rrm1、ido1、egfr、ercc1、her2)的paradigm推断的蛋白质活性(p),这10种蛋白质将经顺铂治疗的肿瘤样品与未经顺铂治疗的肿瘤样品区分开(通过fisher精确检验测得)。蛋白质的“缺乏/失活”活性定义为蛋白质定量在来自>3700个临床样品的所有观察值的最下面的10%中。经由定量质谱法观察为具有在未经顺铂治疗的样品和经顺铂治疗的样品之间有区别的表达水平的大部分蛋白质在经由paradigm分析推断的蛋白质活性方面也呈现相似的有区别的特征。因此,可以使用从同一肿瘤样品测量到的蛋白质活性来交叉检查预测蛋白质活性的途径分析的准确性。
这样获得的基因组突变谱、rna表达谱和任选的蛋白质组分析(从样品测得或通过途径分析推断)可以进一步共同用于鉴定或预测相关信号传导途径中在肿瘤组织中变化最显著的信号传导途径元素。例如,图5a示出了15种蛋白质或蛋白质复合物的热图,其示出了顺铂治疗后的大部分变化。如图所示,预测在顺铂治疗后myc/max和p53活性更有活性,并且预测在顺铂治疗后ets1活性受到最大的抑制。
如此预测的或推断的蛋白质活性可以进一步用于推断信号传导途径(总体上)或甚至包含多个信号传导途径的信号传导网络的活性响应于事件(例如,药物治疗等)如何变化或修饰。换言之,在该途径模型包括单个信号传导途径并且该信号传导途径的唯一阻遏物的推断的蛋白质活性增强的情况下,可以预测该信号传导途径活性(总体上)被下调或抑制。同样,在两个或更多个信号传导途径(其中每个都包括多种信号传导途径元素)的情况下,个体信号传导途径元素的活性可以用于预测在同一途径或受该个体途径元素影响的另一途径中的其他途径元素。例如,图5b示出了多度信号传导网络模型,其中两个或更多个信号传导途径直接或间接连接(例如,ets1途径和flt1途径经由ets1和flt1直接连接,同时ets1途径和flt1途径经由hif1a/arnt复合物或kdr途径间接连接),预测ets1的抑制活性抑制其他信号传导途径或途径元素(包括il-5、flt1、erk1/2、kdr和hif1a/arnt复合物),进一步预测受抑制的其他信号传导途径或途径元素抑制或去抑制其信号传导途径中的下游信号传导途径元素。
诸位发明人进一步设想到了在事件中发生的肿瘤细胞中经修饰的途径活性可以是对该事件的反应的指示。例如,在该事件是对肿瘤细胞的抗肿瘤治疗的情况下,对该事件的反应可以包括增加对抗肿瘤治疗的敏感性或易感性、产生或获得对抗肿瘤治疗的抗性或对抗肿瘤治疗的无反应性。在一些实施例中,可以对在肿瘤细胞中经修饰的途径活性进行评分,以确定对该事件做出反应的可能性。在此类实施例中,可以基于变化的程度(例如,显著增加或减少、适度增加或减少等)、和/或受经修饰的途径活性的影响的下游元素或其他信号传导途径的数目、和/或信号放大的程度(例如,将反馈信号发送到具有经修饰的途径活性的途径元素的下游信号途径元素的数目)、和/或总体活性变化的程度(总的来说,归因于经修饰的途径活性),对每个经修饰的途径活性(例如,每个经修饰的蛋白质表达水平或途径元素的翻译后修饰等)进行评分。当得分高于或低于预定阈值时,这样评分的经修饰的途径活性可能与反应相关(例如,当评分高于阈值时可能获得对抗肿瘤治疗的抗性,并且当评分低于阈值时可能保持对抗肿瘤治疗易感或敏感等)。
进一步设想到了在肿瘤中经修饰的途径活性的推断可以用于预测抗肿瘤治疗对患者的转移性肿瘤的治疗功效。最典型地,如上所述,可以从患者获得来自至少两个不同的解剖位置的多个肿瘤样品,并且组学数据集(例如,基因组学数据、转录组学数据、蛋白质组学数据)可以源自该多个肿瘤样品中的每一个。对于每个肿瘤样品,确定基因组突变谱(例如,多个体细胞突变的突变负荷)和rna表达谱,并且将此类确定的谱整合到途径模型中,以推断每个肿瘤样品中经修饰的途径活性。设想到了从来自两个不同的解剖位置的两个肿瘤样品计算的不同的经修饰的途径活性可能指示可能在转移后肿瘤的经修饰的固有特性,其进一步可能导致对抗肿瘤治疗的敏感性和/或抗性不同。
因此,诸位发明人进一步设想到了基于经修饰的途径活性,并且优选地基于衍生自经修饰的途径活性的得分,可以产生或更新患者的记录,可以推荐新的治疗计划,或者可以更新先前使用的治疗计划。例如,在转移的组织显示出经修饰的途径活性(其指示获得对一种类型的抗肿瘤治疗的抗性)的情况下,患者的记录可以包括给予其他类型的抗肿瘤治疗(例如,免疫疗法、基于quilt的组合疗法等)的信息和/或推荐。
应当理解,本发明主题使用从组学数据集的多个方面获得的综合分子谱,以提高将分子谱与肿瘤组织的生理特性相关联的可信度。此外,将此类多个方面的组学数据集整合,以确定可能影响细胞的固有特性的经修饰的信号传导途径元素、经修饰的信号传导途径活性和经进一步修饰的信号传导途径网络。这种方法允许更准确地预测细胞的固有特性,因为可以总体考虑组学数据多个方面的个体差异。此外,还应当注意,本发明主题仅使用将要用组学数据修饰的相关的信号传导途径模型,这取决于肿瘤的类型和需要获得的信息的类型。这种方法去除了用组学数据进一步过滤从大量的途径分析获得的信息的步骤,以改善至相关数据,使得途径分析的整个数据处理变得快速而且有效。
对于本领域技术人员应当清楚的是,在不脱离本文的发明构思的情况下,除了已经描述的那些之外的更多修改是可能的。因此,除了在所附权利要求的范围中之外,本发明主题不受限制。此外,在解释说明书和权利要求书时,所有术语应当以与上下文一致的尽可能广泛的方式解释。特别地,术语“包含(comprises)”和“包含(comprising)”应当被解释为以非排他性方式指代元素、组件或步骤,从而指示所提及的元素、组件或步骤可以与未明确提及的其他元素、组件或步骤一起存在、或使用、或组合。在说明书权利要求书涉及选自由a、b、c……和n组成的组中的至少一种的情况下,该文字应当被解释为只需要该组中的一个元素,而不是a加n、或b加n等。
- 还没有人留言评论。精彩留言会获得点赞!