热成型工艺后具有增强韧性的热成型膜组合物的制作方法
相关申请的交叉引用本申请要求于2017年9月22日提交的美国临时申请序列号62/561,833的优先权,所述申请的全部公开内容特此通过引用并入。本文所述的实施方案总体上涉及热成型的膜组合物,并且更具体地涉及由于热成型工艺而赋予增强韧性的热成型的膜组合物。
背景技术:
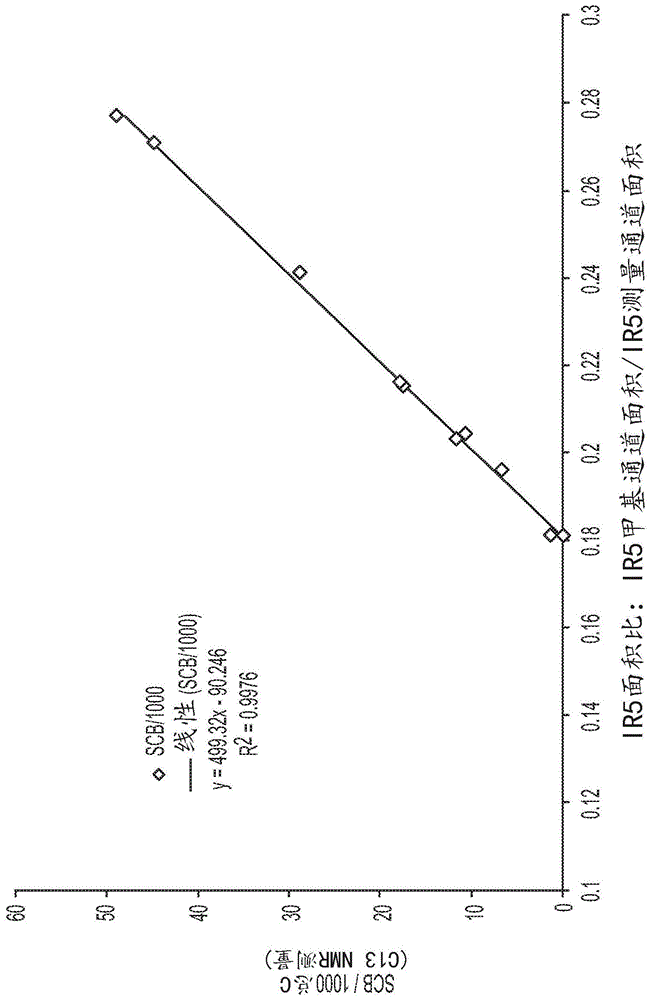
:在许多包装应用中,热成型是最常用的成膜技术之一。在此过程中,将膜加热并在模具中保持固态,使其形成特定形状;然后添加产品,最后将盖膜密封在顶部。热成型膜结构需要满足不同的要求,以完成工艺阶段和最终用途。由于可能包装锋利的产品,因此需要非常好的抗穿刺性。而且,为了容纳重的产品,需要一定的刚度,但是必须与高韧性保持平衡,以保护包装在内部的食品。同样重要的是提高耐低温性,因为许多产品都经过冷冻以保护货架寿命。就是说,由于热成型工艺,传统聚合物组合物的许多韧性均被削弱。因此,需要改进的可热成型组合物,其不仅保持而且改善热成型后的韧性。技术实现要素:本发明的组合物通过在热成型后提供改进的韧性来满足这些需求。在热成型之后,本发明的组合物表现出改善的抗穿刺性、改善的落镖性能和改善的刚度。根据本公开的至少一个实施方案,提供一种热成型的多层膜结构。该热成型多层膜包括芯层和相对于芯层设置在外部的至少两个外层。热成型多层膜结构的至少一层包括第一组合物,其包括至少一种乙烯类聚合物,其中所述第一组合物包括大于0.9的分子加权共聚单体分布指数(mwcdi)值,和满足以下等式的熔体指数比(i10/i2)∶i10/i2≥7.0-1.2xlog(i2)。根据另一个实施方案,提供了一种热成型的单层膜结构。热成型的单层膜结构包含所述第一组合物。由于热成型应用,热成型的单层膜表现出至少10%的抗穿刺性增加。在以下具体实施方式中,这些和其它实施方案更详细地予以描述。附图说明当结合以下附图阅读时,可以最好地理解以下对本公开的具体实施方案的详细描述,在附图中,相似的结构用相似的附图标记指示,并且在附图中:图1描绘了十个scb标准品的“scbf与ir5面积比”的曲线图。图2描绘用于测定样品第一组合物的ir5高度比的几种gpc特征曲线。图3描绘了样品第一组合物的“scbf相对于聚乙烯等效分子logmwi(gpc)”的曲线图。图4描绘样品第一组合物的“共聚单体摩尔%相对于聚乙烯等效logmwi(gpc)”的曲线图。图5是描绘根据本公开的一个或多个实施方案的在热成型之前和之后数个单层膜的抗穿刺性的条形图。图6是条形图,其描绘了根据本公开的一个或多个实施方案的在热成型之前和之后数个多层膜的仪器落镖冲击。图7是描绘根据本公开的一个或多个实施方案的在热成型之前和之后数个多层膜的抗穿刺性的条形图。具体实施方式现在将描述本申请的具体实施方案。但是,本公开可具体化为不同的形式并且不应解释为限于本公开中阐释的实施方案。相反,提供这些实施方案使得本公开将是彻底和完整的,并且将主题的范围充分传递给本领域的技术人员。定义术语“聚合物”是指通过使单体(无论具有相同或不同类型)聚合来制备的聚合化合物。因此,通用术语聚合物涵盖术语“均聚物”,其通常用于指仅由一种类型单体制备的聚合物;以及“共聚物”,其是指由两种或更多种不同单体制备的聚合物。如本文所用,术语“互聚物”是指通过使至少两种不同类型的单体聚合制备的聚合物。因此,通用术语互聚物包括共聚物,以及由多于两种不同类型的单体制备的聚合物,如三元共聚物。“聚乙烯”或“乙烯类聚合物”应意指包含大于50重量%的衍生自乙烯单体的单元的聚合物。此包含聚乙烯均聚物或共聚物(意指衍生自两种或更多种共聚单体的单元)。本领域中已知的聚乙烯的常见形式包含低密度聚乙烯(ldpe);线性低密度聚乙烯(lldpe);超低密度聚乙烯(uldpe);极低密度聚乙烯(vldpe);单位点催化的线性低密度聚乙烯,包含线性和基本上线性低密度树脂(m-lldpe)两种;中密度聚乙烯(mdpe);和高密度聚乙烯(hdpe)。术语“ldpe”也可称为“高压乙烯聚合物”或“高度支化的聚乙烯”,并且定义为意指使用自由基引发剂(如过氧化物)使聚合物在高于14,500psi(100mpa)的压力下在高压釜或管式反应器中部分或完全地均聚或共聚(参见例如us4,599,392,其在此以引用的方式并入)。ldpe树脂通常具有0.916到0.935g/cm范围内的密度。术语“lldpe”包括使用齐格勒-纳塔催化剂体系(ziegler-nattacatalystsystem)制成的树脂以及使用包括但不限于双茂金属催化剂(有时称为“m-lldpe”)和几何形状受限型催化剂的单中心催化剂制成的树脂和使用后茂金属、分子催化剂制成的树脂。lldpe包括线性、基本上线性或非均匀聚乙烯共聚物或均聚物。lldpe比ldpe含有更少的长链支化,并且包括基本上线性乙烯聚合物,其进一步定义于美国专利5,272,236、美国专利5,278,272、美国专利5,582,923和美国专利5,733,155中;均匀支化的线性乙烯聚合物组合物,如美国专利第3,645,992号中的那些;非均匀支化的乙烯聚合物,如根据美国专利第4,076,698号中所公开的方法制备的那些;和/或其共混物(如us3,914,342或us5,854,045中所公开的那些)。可以使用所属领域中已知的任何类型的反应器或反应器配置通过气相、溶液相或浆液聚合或其任何组合来制备lldpe。术语“mdpe”是指密度为0.926至0.935g/cc的聚乙烯。“mdpe”通常使用铬或齐格勒-纳塔催化剂或使用包括但不限于双茂金属催化剂和几何形状受限型催化剂的单中心催化剂来制备。术语“hdpe”是指密度大于约0.935g/cc的聚乙烯,其通常用齐格勒-纳塔催化剂、铬催化剂或单位点催化剂来制备,所述单位点催化剂包括但不限于双茂金属催化剂和限定几何构型的催化剂。术语“uldpe”是指密度为0.880到0.912g/cc的聚乙烯,其通常用齐格勒-纳塔催化剂、包括但不限于双金属茂催化剂和几何形状受限型催化剂的单中心催化剂和后茂金属、分子催化剂制备。如本文所用,术语“丙烯类聚合物”是指以聚合形式包含的聚合物,是指包含大于50重量%的衍生自丙烯单体的单元的聚合物。这包括丙烯均聚物、无规共聚物聚丙烯、抗冲共聚物聚丙烯、丙烯/α-烯烃互聚物和丙烯/α-烯烃共聚物。这些聚丙烯材料在所属领域中众所周知。“多层结构”是指具有多于一层的任何结构。例如,多层结构可以具有二、三、四、五或更多层。可以将多层结构描述为具有以字母表示的层。例如,可以将具有芯层b以及两个外层a和c的三层结构指定为a/b/c。同样,将具有两个芯层b和c以及两个外层a和d的结构指定为a/b/c/d。在一些实施方案中,本发明的多层膜包含多达11个层。现在将详细提及本公开的单层和多层膜结构,其中所述单层和多层膜结构包含第一组合物,所述第一组合物包含至少一种乙烯类聚合物,其中第一组合物包括大于0.9的分子加权共聚单体分布指数(mwcdi)值,并且熔体指数比(i10/i2)满足以下等式:i10/i2≥7.0-1.2×log(i2)。多层膜结构的实施方案包括芯层和相对于芯层在外部设置的至少两个外层。多层膜结构的至少一层包含所述第一组合物。第一组合物可以在多层膜的多层中。例如,第一组合物可以存在于芯层中、在芯层外部的层中或其组合中。第一组合物各种性能有助于改善第一组合物的韧性。例如,与本领域的总密度相同的常规聚合物相比,第一组合物具有优异的共聚单体分布,其在高分子量聚合物分子中共聚单体浓度显著较高,而在低分子量聚合物分子中共聚单体浓度显著较低。还已经发现,与常规聚合物相比,第一组合物具有较低的lcb(长链分支),如低的zsvr所指示。由于共聚单体的这种分布以及低lcb性质,第一组合物具有更多的连接链,并且因此改善了膜韧性。如上所述,第一组合物包括大于0.9的mwcdi值。在一个实施方案中,第一组合物的mwcdi值小于或等于10.0,进一步小于或等于8.0,进一步小于或等于6.0。在另一个实施方案中,第一组合物的mwcdi值小于或等于5.0,进一步小于或等于4.0,进一步小于或等于3.0。在又一个实施方案中,第一组合物的mwcdi值大于或等于1.0,进一步大于或等于1.1,进一步大于或等于1.2。在其它实施方案中,第一组合物的mwcdi值大于或等于1.3,进一步大于或等于1.4,进一步大于或等于1.5。第一组合物的熔体指数比(i10/i2)满足以下等式:i10/i2≥7.0-1.2xlog(i2)。在又一个实施方案中,第一组合物的熔体指数比i10/i2大于或等于7.0,进一步大于或等于7.1,进一步大于或等于7.2,进一步大于或等于7.3。在一个实施方案中,第一组合物的熔体指数比i10/i2小于或等于9.2,进一步小于或等于9.0,进一步小于或等于8.8,进一步小于或等于8.5。在一个实施方案中,第一组合物的zsvr值为1.2到3.0,或1.2到2.5,或1.2到2.0。在又一个实施方案中,第一组合物的乙烯基不饱和度含量每1,000,000个总碳大于10个乙烯基。举例来说,每1,000,000个总碳大于20个乙烯基,或每1,000,000个总碳大于50个乙烯基,或每1,000,000个总碳大于70个乙烯基,或每1,000,000个总碳大于100个乙烯基。使用以下定义的核磁共振(nmr)光谱计算乙烯基不饱和度。在一个实施方案中,第一组合物的密度在0.900g/cc到0.960g/cm3、或0.910到0.940g/cm3、或0.910到0.930、或0.910到0.925g/cm3范围内。例如,密度可以是从0.910、0.912或0.914g/cm3的下限至0.925、0.927或0.930g/cm3的上限(1cm3=1cc)。在进一步实施方案中,第一组合物的熔体指数(i2;在190℃/2.16kg下)为0.1至50克/10分钟,例如0.1至30克/10分钟,或0.1至20克/10分钟,或0.1至10克/10分钟。举例来说,熔体指数(i2;在190℃/2.16kg下)可为从下限0.1、0.2或0.5g/10分钟到上限1.0、2.0、3.0、4.0、5.0、10、15、20、25、30、40或50g/10分钟。在另一个实施方案中,第一组合物的分子量分布表示为如通过常规凝胶渗透色谱法(gelpermeationchromatography,gpc)(conv.gpc)所测定的重均分子量与数均分子量(mw/mn)的比率,在2.2至5.0范围内。举例来说,分子量分布(mw/mn)可从下限2.2、2.3、2.4、2.5、3.0、3.2或3.4到上限3.9、4.0、4.1、4.2、4.5或5.0。在一个实施方案中,第一组合物的数均分子量(mn)如通过conv.gpc测定在10,000到50,000g/mol范围内。举例来说,数均分子量可从下限10,000、20,000或25,000g/mol到上限35,000、40,000、45,000或50,000g/mol。在另一个实施方案中,乙烯类聚合物的重均分子量(mw)如通过conv.gpc测定在70,000至200,000g/mol范围内。例如,数均分子量可从下限70,000、75,000或78,000克/mol到上限120,000、140,000、160,000、180,000或200,000g/mol。在一个实施方案中,第一组合物的熔融粘度比eta*0.1/eta*100在2.2到7.0范围内,其中eta*0.1是在0.1rad/s的剪切速率下计算的动态粘度,并且eta*100是在100rad/s的剪切速率下计算的动态粘度。以下提供了有关熔融粘度比和动态粘度计算的其它细节。在一个实施方案中,第一组合物的乙烯类聚合物为乙烯/α-烯烃互聚物,并且进一步为乙烯/α-烯烃共聚物。α-烯烃可具有小于或等于20个碳原子。例如,α-烯烃共聚单体可具有3到10个碳原子,或3到8个碳原子。示例性α-烯烃共聚单体包含(但不限于)丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯、1-癸烯和4-甲基-1-戊烯。一种或多种α-烯烃共聚单体可例如选自以下组成的组:丙烯、1-丁烯、1-己烯和1-辛烯;或在替代方案中,选自以下组成的组:1-丁烯、1-己烯和1-辛烯,并且进一步1-己烯和1-辛烯。乙烯类聚合物可包括小于20重量%的衍生自一种或多种α-烯烃共聚单体的单元。小于18重量%的所有个别值和子范围都包括在本文中并公开于本文中;举例来说,乙烯类聚合物可包含小于15重量%的衍生自一种或多种α-烯烃共聚单体的单元;或在替代方案中,小于10重量%的衍生自一种或多种α-烯烃共聚单体的单元;或在替代方案中,1到20重量%的衍生自一种或多种α-烯烃共聚单体的单元;或在替代方案中,1到10重量%的衍生自一种或多种α-烯烃共聚单体的单元。相反,乙烯类聚合物可包括至少80重量%的衍生自乙烯的单元。至少80重量%的所有个别值和子范围都包括在本文中并公开于本文中;举例来说,乙烯类聚合物可包括至少82重量%的衍生自乙烯的单元;或在替代方案中,至少85重量%的衍生自乙烯的单元;或在替代方案中,至少90重量%的衍生自乙烯的单元;或在替代方案中,80到100重量%的衍生自乙烯的单元;或在替代方案中,90到100重量%的衍生自乙烯的单元。任选地,第一组合物进一步可包括第二乙烯类聚合物。在其它实施方案中,第二乙烯类聚合物为乙烯/α-烯烃互聚物,并且进一步为乙烯/α-烯烃共聚物或ldpe。合适的α-烯烃共聚单体在上面列出。在一个实施方案中,第二乙烯类聚合物为非均匀支化的乙烯/α-烯烃互聚物,并且进一步为非均匀支化的乙烯/α-烯烃共聚物。非均匀支化的乙烯/α-烯烃互聚物和共聚物通常使用齐格勒/纳塔(ziegler/natta)型催化剂体系来产生,并且更多的共聚单体分布在聚合物的较低分子量分子中。在一个实施方案中,第二乙烯类聚合物的分子量分布(mw/mn)在3.0至5.0的范围内,例如3.2至4.6。例如,分子量分布(mw/mn)可以是3.2、3.3、3.5、3.7或3.9的下限至4.6、4.7、4.8、4.9或5.0的上限。在一个实施方案中,该组合物还包含另一种聚合物。在其它实施方案中,聚合物选自以下:lldpe、mdpe、ldpe、hdpe、丙烯类聚合物或其组合。在一个实施方案中,第一组合物还包含ldpe。在其它实施方案中,以组合物的重量计,ldpe以5到50重量%,进一步10到40重量%,进一步15到30重量%的量存在。在其它实施方案中,ldpe的密度为0.915到0.925g/cc,并且熔体指数(i2)为0.5到5g/10min,进一步为1.0到3.0g/10min。在其它实施方案中,第一组合物可包括一种或多种添加剂。添加剂包含(但不限于)抗静电剂、增色剂、染料、润滑剂、填充剂(例如tio2或caco3)、遮光剂、成核剂、加工助剂、颜料、主抗氧化剂、次抗氧化剂、uv稳定剂、防结块剂、助滑剂、增粘剂、阻燃剂、抗微生物剂、消味剂、抗真菌剂和其组合。热成型膜的其他成分另外,在一个或多个实施方案中,热成型多层膜可包含丙烯类聚合物。各种丙烯类聚合物产品被认为是合适的,例如,丙烯-乙烯共聚物树脂,如versifytm3000或3200,它们均由密歇根州米德兰市的陶氏化学公司提供。在一些实施方案中,热成型的多层膜结构基本上不含丙烯类聚合物。如本文所用,“基本上不含丙烯类聚合物”是指小于热成型多层膜的0.5重量%,或小于0.1重量%,或小于0.01重量%。此外,在进一步的实施方案中,热成型的单层或多层膜结构基本上由乙烯类聚合物组成。如本文所用,“基本上由......组成”是指热成型的单层或多层膜结构可包括其他添加剂,但限于乙烯类聚合物。除了第一组合物的乙烯类聚合物之外,热成型的单层或多层膜结构还可包括其他乙烯类聚合物。在一个实施方案中,当在2.16kg的负载和190℃的温度下根据astmd1238测量时,热成型的多层膜结构可包含密度为0.895至0.965g/cm3且熔体指数(i2)为0.5至6.0g/10min的乙烯类聚合物。在进一步的实施方案中,密度可以为0.900至0.940g/cm3,或0.900至0.925g/cm3,并且熔体指数(i2)为0.85至3.5g/10min,或2.0至6.0g/10min。实例包括但不限于dowtm低密度聚乙烯(ldpe)和线性低密度聚乙烯(lldpe)、dowlextm线性低密度聚乙烯(lldpe)、attanetm超低密度聚乙烯(uldpe)、elitetm增强聚乙烯,每种都可以从密歇根州米德兰市的陶氏化学公司购得。在进一步的实施方案中,热成型的多层膜可在一个或多个层中包含另外的组合物。在一些实施方案中,本公开的多层膜可以包含一个或多个阻隔层。在这类实施方案中,阻隔层可以包含一种或多种聚酰胺(尼龙)、乙烯乙烯醇共聚物(evoh)和/或马来酸化聚烯烃。evoh可以包括具有27到44mol%乙烯的乙烯醇共聚物,并且是通过例如水解乙酸乙烯酯共聚物来制备。可用于本发明的实施方案的市售evoh的实例包括可乐丽(kuraray)的evaltm和nippongoshei的noltextm。在阻挡层包含聚酰胺的实施方案中,聚酰胺可以包括聚酰胺6、聚酰胺9、聚酰胺10、聚酰胺11、聚酰胺12、聚酰胺6,6、聚酰胺6/66和如聚酰胺61、聚酰胺6t、mxd6的芳香族聚酰胺或其组合。可以将马来酸化聚烯烃用于热成型多层膜的各个层。在一些实施方案中,马来酸化聚烯烃可以在热成型的多层膜的结合层中。马来酸化聚烯烃可包括马来酸酐接枝聚乙烯或马来酸酐接枝聚丙烯。马来酸酐接枝的聚乙烯的合适的商业实例是购自陶氏化学公司(密歇根州密德兰)的amplifytmty1057h。在其他实施方案中,多层或单层膜可以不含聚酰胺。在一些这样的实施方案中,与包含聚酰胺的多层膜相比,不存在聚酰胺可以引起具有热成型特性的较便宜的多层膜。在一些实施方案中,多层膜可以包含小于5重量%聚酰胺或小于3重量%聚酰胺或小于1重量%聚酰胺或小于0.5重量%聚酰胺,各者都以膜的总重量计。然而,如上文所指示,存在一些实施方案(例如当聚酰胺将用作阻隔层时),其中多层膜包括更大量的聚酰胺。在一些实施方案中,多层膜可以包括结合层。结合层可以用于在共挤出期间将两个层粘附在一起,特别是在两个层的组合物之间存在不相容性的情况下。举例来说,如果多层膜包含乙烯乙烯醇阻隔层,则可以使用结合层将乙烯乙烯醇层粘附到主要包含聚烯烃的层上。所属领域的技术人员可以确定是否需要结合层,并且如果是这样的话,则基于本文的教示内容,视待包括在多层膜中的层的组成而定来选择适当的结合层。应理解本发明的多层膜内部的层中的任一层可以进一步包含如所属领域的技术人员已知的一种或多种添加剂,例如抗氧化剂、紫外线稳定剂、热稳定剂、助滑剂、防结块剂、颜料或着色剂、加工助剂、交联催化剂、阻燃剂、填充剂和发泡剂。本公开的单层或多层膜的总厚度(热成型前)优选在10μm到250μm、,优选50μm到200μm或100μm到180μm范围内。单独层厚度可以取决于可用层的数量、层的类型(例如表层、阻隔层、内层等、)和膜的总厚度而变化。在一些实施方案中,可以使用所属领域的技术人员已知的技术来共挤出本公开的多层膜(例如使用吹塑膜工艺或流延膜工艺、)。在一些实施方案中,本公开的多层坍缩的或单层膜非常适合用于热成型应用。在一个或多个实施方案中,基于第一组合物的总多层膜厚度的百分比,多层膜可占至少50%。在另一个实施方案中,基于第一组合物的总多层膜厚度的百分比,多层膜可占至少60%。可以使用所属领域的技术人员已知的技术将本多层和单层膜成型为各种制品。例如,在一些实施方案中,可以将单层和多层膜热成型为制品。这类制品的实例包括刚性容器、柔性托盘和半柔性包装。这类制品可以用于例如包装食品中,如水果、奶酪、肉、加工肉、加工食品和冷冻食品。单层和多层膜的性质由于热成型,热成型的单层膜可以显示出至少10%的抗穿刺性的增加。在进一步的实施方案中,由于热成型步骤,热成型的单层膜可显示出至少15%或至少20%的抗穿刺性的增加。另外,热成型的聚烯烃多层膜结构也可以表现出改善的性能。例如,当根据astmd3763测量时,热成型的聚烯烃多层膜可具有至少0.6j、或至少0.8j、或至少1.0j的仪器落镖冲击。制备第一组合物的聚合方法为了生产第一组合物的乙烯类聚合物,合适的聚合方法可包括但不限于使用一个或多个常规反应器(例如环管反应器、等温反应器、绝热反应器、搅拌槽反应器、并联、串联的高压釜反应器和/或其任何组合)的溶液聚合方法。乙烯类聚合物组合物可以例如通过溶液相聚合方法,使用一个或多个环管反应器、绝热反应器和其组合来产生。一般来说,溶液相聚合方法在一个或多个充分混合的反应器(如一个或多个环管反应器和/或一个或多个绝热反应器)中在115至250℃(例如135至200℃)范围内的温度下,并且在300至1000psig(例如450至750psig)范围内的压力下进行。在一个实施方案中,乙烯类聚合物可在串联配置的两个环管反应器中生产,第一反应器温度在115℃到200℃,例如135℃到165℃的范围内,并且第二反应器温度在150℃到210℃,例如185℃到200℃的范围内。在另一实施方案中,乙烯类聚合物组合物可以在单个反应器中产生,反应器温度在115℃到200℃,例如130℃到190℃的范围内。溶液相聚合方法中的滞留时间通常在2到40分钟,例如5到20分钟的范围内。将乙烯、溶剂、一种或多种催化剂系统、任选地一种或多种助催化剂和任选地一种或多种共聚单体连续地进料到一个或多个反应器中。示例性溶剂包含(但不限于)异烷烃。举例来说,这类溶剂可以名称isopare商购自埃克森美孚化工(exxonmobilchemical)。随后从一个或多个反应器中去除乙烯类聚合物组合物与溶剂的所得混合物,并且分离乙烯类聚合物组合物。溶剂通常通过溶剂回收单元(即,热交换器和分离器容器)回收,并且随后再循环回到聚合系统中。在一个实施方案中,第一组合物的乙烯类聚合物可通过溶液聚合方法在双反应器系统(例如双环管反应器系统)中产生,其中乙烯和任选的一种或多种α-烯烃在存在一种或多种催化剂系统下在一个反应器中聚合以产生第一乙烯类聚合物,并且乙烯和任选的一种或多种α-烯烃在存在一种或多种催化剂系统下在第二反应器中聚合以产生第二乙烯类聚合物。另外,可存在一种或多种助催化剂。在另一个实施方案中,乙烯类聚合物可通过溶液聚合方法在单个反应器系统(例如单环管反应器系统)中产生,其中乙烯和任选的一种或多种α-烯烃在存在一种或多种催化剂系统下聚合。另外,可存在一种或多种助催化剂。如上所述,本发明提供了一种形成包含至少两种乙烯类聚合物的组合物的方法,所述方法包括以下步骤:在包含结构i的金属-配体络合物的催化剂系统的存在下,在溶液中聚合乙烯和任选的至少一种共聚单体以形成第一乙烯类聚合物;并且在包含齐格勒/纳塔催化剂的催化剂系统的存在下聚合乙烯和任选的至少一种共聚单体以形成第二乙烯类聚合物;并且其中结构i如下:其中:m是钛、锆或铪,其各自独立地处于+2、+3或+4的形式氧化态;并且n是0到3的整数,并且其中当n是0时,x不存在;并且每个x独立地为中性、单阴离子或双阴离子的单齿配体;或两个x连在一起以形成中性、单阴离子或双阴离子的双齿配体;并且x和n以使得式(i)的金属-配体络合物整体上呈中性的方式选择;以及每个z独立地为o、s、n(c1-c40)烃基或p(c1-c40)烃基;并且其中z-l-z片段包括式(1):r1至r16各自独立地选自以下组成的组群:经取代或未经取代的(c1-c40)烃基、经取代或未经取代的(c1-c40)杂烃基、si(rc)3、ge(rc)3、p(rp)2、n(rn)2、orc、src、no2、cn、cf3、rcs(o)-、rcs(o)2-、(rc)2c=n-、rcc(o)o-、rcoc(o)-、rcc(o)n(r)-、(rc)2nc(o)-、卤素原子、氢原子;并且其中每个rc独立地是(c1-c30)烃基;rp是(c1-c30)烃基;并且rn是(c1-c30)烃基;并且其中,任选地,两个或更多个r基团(从r1到r16)可以一起结合成一个或多个环结构,这些环结构各自独立地在环中具有3到50个原子,不包括任何氢原子。本方法可以包含如本文所描述的两个或更多个实施方案的组合。在一个实施方案中,所述方法包括在溶液中,在包括结构i的金属-配位体络合物的催化剂系统存在下乙烯和任选的至少一种α-烯烃聚合,以形成第一乙烯类聚合物;并且在包括齐格勒/纳塔催化剂的催化剂系统存在下乙烯和任选的至少一种α-烯烃聚合,以形成第二乙烯类聚合物。在其它实施方案中,每个α-烯烃独立地为c1-c8α-烯烃。在一个实施方案中,r9至r13或r4至r8中任选地两个或更多个r基团可一起组合成一个或多个环结构,这类环结构在环中各自独立地具有3到50个原子,不包含任何氢原子。在一个实施方案中,m是铪。在一个实施方案中,r3和r14各自独立地为烷基,并且进一步为c1-c3烷基,并且进一步为甲基。在一个实施方案中,r1和r16各自如下:在一个实施方案中,芳基、杂芳基、烃基、杂烃基、si(rc)3、ge(rc)3、p(rp)2、n(rn)2、orc、src、rcs(o)-、rcs(o)2-、(rc)2c=n-、rcc(o)o-、rcoc(o)-、rcc(o)n(r)-、(rc)2nc(o)-、亚烃基和杂亚烃基中的每一个独立地为未经取代的或经一个或多个rs取代基取代;并且每个rs独立地为卤原子、多氟取代、全氟取代、未经取代的(c1-c18)烷基、f3c-、fch2o-、f2hco-、f3co-、r3si-、r3ge-、ro-、rs-、rs(o)-、rs(o)2-、r2p-、r2n-、r2c=n-、nc-、rc(o)o-、roc(o)-、rc(o)n(r)-或r2nc(o)-,或两个rs一起形成未经取代的(c1-c18)亚烷基,其中每个r独立地为未经取代的(c1-c18)烷基。在一个实施方案中,r1至r16中的两个或更多个不会组合形成一个或多个环结构。在一个实施方案中,适用于产生第一乙烯/α-烯烃互聚物的催化剂系统是包括由以下结构表示的双((2-氧代基-3-(二苯并-1h-吡咯-1-基)-5-(甲基)苯基)-2-苯氧基甲基)-亚甲基-1,2-环己二基铪(iv)二甲基的催化剂系统:ia:适合于本发明中的齐格勒/纳塔催化剂为典型的负载型齐格勒型催化剂,其在溶液方法的高聚合温度下特别有用。这类组合物的实例是衍生自有机镁化合物、烷基卤化物或铝卤化物或氯化氢和过渡金属化合物的组合物。这类催化剂的实例描述于美国专利第4,612,300号;第4,314,912号以及第4,547,475号中,所述专利的教导内容以引用的方式并入本文中。具体地,合适的有机镁化合物包含例如烃类可溶的二烃基镁,如二烷基镁和二芳基镁。示例性合适的二烷基镁具体地包含正丁基-仲丁基镁、二异丙基镁、二正己基镁、异丙基-正丁基镁、乙基-正己基镁、乙基-正丁基镁、二正辛基镁等,其中烷基具有1到20个碳原子。示例性适合二芳基镁包含二苯基镁、二苄基镁和二甲苯基镁。合适的有机镁化合物包含烷基镁烷氧化物与烷基镁芳基氧化物和芳基镁烷氧化物与芳基镁芳基氧化物,以及芳基镁卤化物和烷基镁卤化物,其中不含卤素的有机镁化合物是更期望的。卤化物源包含活性非金属卤化物、金属卤化物和氯化氢。合适的非金属卤化物由式r'x表示,其中r′为氢或活性一价有机基团,并且x为卤素。特别合适的非金属卤化物包含(例如),氢卤化物和活性有机卤化物,如叔烷基卤化物、烯丙基卤化物、苄基卤化物和其它活性烃基卤化物。通过活性有机卤化物意指含有不稳定卤素的烃基卤化物,所述不稳定卤素至少与仲丁基氯的卤素活性相同(即容易丢失给另一化合物),优选与叔丁基氯活性相同。除了有机单卤化物之外,应理解,还可以适合地利用具有如上所定义的活性的有机二卤化物、三卤化物和其它多卤化物。优选的活性非金属卤化物的实例包含氯化氢、溴化氢、叔丁基氯、叔戊基溴、烯丙基氯、苄基氯、巴豆基氯、甲基乙烯基碳酰氯、α-苯基乙基溴、二苯甲基氯等。最优选为氯化氢、叔丁基氯、烯丙基氯以及苄基氯。合适的金属卤化物包含由式mry-axa表示的那些金属卤化物,其中:m为门捷列夫元素周期表的第iib族、第iiia族或第iva族的金属;r为一价有机基团;x为卤素;y具有对应于m的价态的值;并且“a”的值为1到y。优选的金属卤化物为式alr3-axa的铝卤化物,其中每个r独立地为烃基,如烷基;x为卤素并且a为1到3的数。最优选是烷基铝卤化物,如乙基倍半氯化铝、二乙基氯化铝、乙基二氯化铝和二乙基溴化铝,乙基二氯化铝是尤其优选的。替代地,可合适地使用金属卤化物如三氯化铝或三氯化铝与烷基铝卤化物的组合或三烷基铝化合物。常规齐格勒-纳塔过渡金属化合物中的任一者可有用地用作制备负载型催化剂组分中的过渡金属组分。通常,过渡金属组分是第ivb族、第vb族或第vib族金属的化合物。过渡金属组分通常由下式表示:trx'4-q(or1)q、trx'4-q(r2)q、vox'3和vo(or)3。tr是第ivb族、第vb族或第vib族金属,优选是第ivb族或第vb族金属,优选是钛、钒或锆;q是0或等于或小于4的数;x′是卤素,并且r1是具有1到20个碳原子的烷基、芳基或环烷基;并且r2是烷基、芳基、芳烷基、被取代的芳烷基等。芳基、芳烷基和经取代的芳烷基含有1到20个碳原子,优选1到10个碳原子。当过渡金属化合物含有烃基r2(为烷基、环烷基、芳基或芳烷基)时,在β到金属碳键的位置中烃基将优选不含有h原子。芳烷基的说明性但非限制性实例为甲基、新戊基、2,2-二甲基丁基、2,2-二甲基己基;芳基如苄基;环烷基如1-降冰片烷基。如果需要,可以使用这些过渡金属化合物的混合物。过渡金属化合物的说明性实例包括ticl4、tibr4、ti(oc2h5)3cl、ti(oc2h5)cl3、ti(oc4h9)3cl、ti(oc3h7)2cl2、ti(oc6h13)2cl2、ti(oc8h17)2br2和ti(oc12h25)cl3、ti(o-ic3h7)4和ti(o-nc4h9)4。钒化合物的说明性实例包括vcl4、vocl3、vo(oc2h5)3和vo(oc4h9)3。锆化合物的说明性实例包含zrcl4、zrcl3(oc2h5)、zrcl2(oc2h5)2、zrcl(oc2h5)3、zr(oc2h5)4、zrcl3(oc4h9)、zrcl2(oc4h9)2以及zrcl(oc4h9)3。无机氧化物载体可以用于催化剂的制备,并且载体可以是任何粒状氧化物或混合的氧化物,所述混合氧化物已经热或化学脱水使得其基本上不含吸附水分。参见美国专利第4,612,300号;第4,314,912号以及第4,547,475号;其教导内容以引入的方式并入本文中。上文所描述的催化剂系统可以通过使其与活化共催化剂接触、或使其与活化共催化剂组合,或通过使用活化技术(例如本领域中已知用于基于金属的烯烃聚合反应的活化技术)而显现为催化活性。用于本文的适合活化助催化剂包含烷基铝;聚合或寡聚铝氧烷(alumoxane)(也称为铝氧烷(aluminoxane));中性路易斯酸;和非聚合、非配位、形成离子的化合物(包含这类化合物在氧化条件下的使用)。一种合适的活化技术是本体电解(bulkelectrolysis)。还涵盖前述活化助催化剂和技术的一种或多种的组合。术语“烷基铝”意指单烷基铝二氢化物或单烷基铝二卤化物、二烷基氢化铝或二烷基铝卤化物或三烷基铝。铝氧烷及其制剂在例如美国专利6,103,657中是已知的。优选的聚合或寡聚铝氧烷的实例是甲基铝氧烷、三异丁基铝改性的甲基铝氧烷及异丁基铝氧烷。示例性路易斯酸活化助催化剂为含有1至3个如本文所述的烃基取代基的第13族金属化合物。在一些实施方案中,示例性第13族金属化合物是三(烃基)取代的铝化合物或三(烃基)-硼化合物。在一些其它实施方案中,示例性第13族金属化合物是三(烃基)取代的铝化合物或三(烃基)-硼化合物是三((c1-c10)烷基)铝或三((c6-c18)芳基)硼化合物以及其卤化(包含全卤化)衍生物。在一些其它实施方案中,示例性第13族金属化合物是三(氟取代的苯基)硼烷,在其它实施方案中,是三(五氟苯基)硼烷。在一些实施方案中,活化助催化剂为三((c1-c20)烃基)硼酸酯(例如四氟硼酸三苯甲酯)或三((c1-c20)烃基)铵四((c1-c20)烃基)硼烷(例如双(十八基)甲基铵四(五氟苯基)硼烷)。如本文所用,术语“铵”意指氮阳离子,其为((c1-c20)烃基)4n+、((c1-c20)烃基)3n(h)+、((c1-c20)烃基)2n(h)2+、(c1-c20)烃基n(h)3+或n(h)4+,其中每个(c1-c20)烃基可以相同或不同。中性路易斯酸活化助催化剂的示例性组合包含包括三((c1-c4)烷基)铝和卤化三((c6-c18)芳香基)硼化合物,尤其是三(五氟苯基)硼烷的组合的混合物。其它示例性实施方案是这类中性路易斯酸混合物与聚合或寡聚铝氧烷的组合,和单一中性路易斯酸、尤其是三(五氟苯基)硼烷与聚合或寡聚铝氧烷的组合。(金属-配体络合物):(三(五氟-苯基硼烷):(铝氧烷)[例如,(第4族金属-配体络合物):(三(五氟-苯基硼烷):(铝氧烷)]的示例性实施方案摩尔数之比为1∶1∶1到1∶10∶30,其它示例性实施方案为1∶1∶1.5到1∶5∶10。先前在以下uspn中已经关于不同金属-配体络合物教导许多活化助催化剂和活化技术:us5,064,802;us5,153,157;us5,296,433;us5,321,106;us5,350,723;us5,425,872;us5,625,087;us5,721,185;us5,783,512;us5,883,204;us5,919,983;us6,696,379;和us7,163,907。合适的烃基氧化物的实例公开于us5,296,433中。适用于加成聚合催化剂的布朗斯特酸(bronstedacid)盐的实例公开于us5,064,802、us5,919,983、us5,783,512中。适用作加成聚合催化剂的活化助催化剂的阳离子氧化剂和非配位相容性阴离子的盐的实例公开于us5,321,106中。适用作加成聚合催化剂的活化助催化剂的碳离子盐的实例公开于us5,350,723中。适用作加成聚合催化剂的活化助催化剂的硅烷基盐的实例公开于us5,625,087中。醇、硫醇、硅醇和肟与三(五氟苯基)硼烷的合适的络合物的实例公开于us5,296,433中。这些催化剂中的一些还描述于us6,515,155b1的一部分中,在第50栏第39行开始直到第56栏第55行,仅其中所述部分以引用的方式并入本文中。在一些实施方案中,上述催化剂系统可通过与一种或多种助催化剂(如阳离子形成助催化剂、强路易斯酸或其组合)组合而活化以形成活性催化剂组合物。适用的助催化剂包含聚合或寡聚铝氧烷,特别是甲基铝氧烷,以及惰性相容性非配位离子形成化合物。示例性合适的助催化剂包含(但不限于)改性的甲基铝氧烷(mmao)、双(氢化牛脂烷基)甲基、四(五氟苯基)硼酸盐(1-)胺、三乙基铝(tea)和其任何组合。在一些实施方案中,一种或多种前述活化助催化剂彼此组合使用。在一个实施方案中,可使用三((c1-c4)烃基)铝、三((c1-c4)烃基)硼烷或具有寡聚或聚合铝氧烷化合物的硼酸铵的混合物的组合。测试方法测试方法包含以下:熔体指数(i2)根据astmd-1238在190℃下于2.16kg下测量熔体指数(i2)。数值以克/10分钟为单位报告,其与每10分钟洗脱的克数相对应。密度根据astmd4703制备用于密度测量的样品,并且以克/立方厘米(g/cc或g/cm3)为单位报告。测量是在使用astmd792方法b按压样品一小时内进行的。动态剪切流变学每个样品在177℃、10mpa压力下,在空气中压缩成型为“3mm厚×25mm直径”圆形薄片维持五分钟。随后将样品从压机中取出,并且放在柜台面冷却。恒温频率扫描测量在氮气吹扫下,在配备有25mm平行板的ares应变受控流变仪(tainstruments)上进行。对于每次测量,在间隙归零之前,将流变仪热平衡至少30分钟。将样品盘放在板上,并且使其在190℃下熔融五分钟。随后使板靠近为2mm,修整样品,并且随后开始测试。所述方法另外内设五分钟延迟,以允许温度平衡。实验是在190℃下经0.1rad/s到100rad/s的频率范围以每十秒时间间隔五个点进行。应变幅度恒定在10%。根据幅度和相位分析压力反应,由其计算储能模量(g′)、损耗模量(g″)、复模量(g*)、动态粘度(η*或eta*)和tanδ(或tandelta)。熔融强度在连接于高特福里欧特斯特(gottfertrheotester)2000毛细管流变仪的高特福里欧滕斯(gottfertrheotens)71.97(南卡罗莱纳州石山的高特福有限公司(inc.;rockhill,sc))上进行熔融强度测量。将聚合物熔融物以平的进入角(180度)挤压通过毛细管模,毛细管直径是2.0mm并且纵横比(毛细管长度/毛细管直径)是15。样品在190℃下平衡10分钟之后,活塞以0.265毫米/秒的恒定活塞速度运行。标准测试温度是190℃。将样品(约20克)以2.4mm/s2的加速度单向地抽拉到位于模具下方100mm的一组加速轧缝。随轧辊的卷绕速度变化记录下拉力。熔融强度报告为在链断裂前的平线区力(cn)。在熔融强度测量中使用以下条件:柱塞速度=0.265mm/s;轮盘加速度=2.4mm/s2;毛细管直径=2.0mm;毛细管长度=30mm;并且筒管直径=12mm。常规凝胶渗透色谱法(conv.gpc)来自珀里莫查公司(polymerchar)(西班牙的巴伦西亚(valencia,spain))的gpc-ir高温色谱系统配备有精密检测器(马萨诸塞州的阿姆赫斯特(amherst,ma))、2040型2-角度激光散射检测器、ir5红外检测器和4-毛细管粘度计(均来自珀里莫查公司)。使用珀里莫查公司的仪器控制软件和数据采集界面进行数据采集。系统配备有来自安捷伦技术公司(加利福尼亚州圣克拉拉)的在线溶剂脱气装置和泵送系统。注射温度控制在150℃。使用的柱为来自聚合物实验室(polymerlaboratories)(英国什罗普郡(shropshire,uk))的三个10微米“混合b”柱。使用的溶剂是1,2,4-三氯苯。样品以“50毫升溶剂中0.1克聚合物”的浓度制备。色谱溶剂和样品制备溶剂各自含有“200ppm的丁基化羟基甲苯(bht)”。两种溶剂源都经氮气鼓泡。乙烯类聚合物样品在160℃下温和地搅拌三小时。注射体积为“200微升”,并且流动速率为“1毫升/分钟。”gpc柱组通过运行21种“窄分子量分布”聚苯乙烯标准品来进行校准。标准品的分子量(mw)在580g/mol到8,400,000g/mol范围内,并且所述标准品含于六种“混合液(cocktail)”混合物中。每种标准混合物在个别分子量之间具有至少十倍间隔。标准混合物购自聚合物实验室。聚苯乙烯标准样品针对等于或大于1,000,000g/mole的分子量以“50ml溶剂中0.025g”制备,并且针对小于1,000,000g/mole的分子量以“50ml溶剂中0.050g”制备。聚苯乙烯标准物在轻缓搅动下,在80℃下溶解30分钟。首先运行窄标准物混合物,并且按照“最高分子量组分”递减的次序最小化降解。使用等式1将聚苯乙烯标准品峰值分子量转换为聚乙烯分子量(如williams和ward,j.polym.sci.,《聚合物快报(polym.let.)》,6,621(1968)中所述):m聚乙烯=a×(m聚苯乙烯)b(等式1),其中m是分子量,a等于0.4316并且b等于1.0。根据以下等式2-4计算数均分子量(mn(convgpc))、重均分子量(mw-convgpc)和z均分子量(mz(convgpc)):在等式2-4中,rv是柱保留体积(线性间隔),以“每秒1点”收集,ir为来自gpc仪器的ir5测量通道的减去基线的ir检测器信号,以伏为单位,并且mpe为由等式1测定的聚乙烯当量mw。用来自珀里莫查公司的“gpcone软件(2.013h版)”进行数据计算。蠕变零剪切粘度测量方法零剪切粘度是经由蠕变测试获得,所述蠕变测试是在190℃使用“25mm直径”的平行板在ar-g2应力控制流变仪(tainstruments;特拉华州纽卡斯尔(newcastle,del))进行。在装置归零之前,将流变仪烘箱设定成测试温度,保持至少30分钟。在测试温度下,将压缩成型样品盘插入板间,并且使其平衡五分钟。上部板随后降低到比所期望的测试间隙(1.5mm)高50μm(仪器设定)。修整掉任何多余的材料,并且将上部板下降到所要间隙。在流动速率为5l/min的氮气吹扫下进行测量。默认蠕变时间设定为两个小时。每个样品在177℃下,在空气中在10mpa压力下,持续五分钟压缩模制成“2mm厚×25mm直径”圆形板。随后将样品从压机中取出,并且放在柜台面冷却。对所有样品施加20pa的恒定低剪应力以确保稳定状态的剪切速率低到足以处于牛顿区(newtonianregion)。对于这一研究中的样品,所得稳定状态剪切速率在10-3到10-4s-1范围内。稳定状态由“log(j(t))对log(t)”曲线的最后10%时间窗口中的所有数据进行线性回归来确定,其中j(t)为蠕变柔量并且t为蠕变时间。如果线性回归的斜率大于0.97,那么认为达到稳定状态,随后停止蠕变测试。在此研究的所有情况下,斜率在一小时内满足所述标准。稳定状态剪切速率由“ε对t”曲线的最后10%时间窗口中的所有数据点进行线性回归的斜率来确定,其中ε是应变。零剪切粘度由施加的应力与稳定状态的剪切速率的比率确定。为了确定样品在蠕变测试期间是否降解,在蠕变测试之前和之后从0.1至100弧度/秒对相同样本进行小振幅振荡剪切测试。比较两个测试的复数粘度值。如果在0.1弧度/秒下的粘度值的差异大于5%,那么认为样品在蠕变测试期间已降解,并且舍弃结果。零剪切粘度比(zsvr)定义为根据以下等式5,在当量重均分子量(mw(convgpc))下,分支聚乙烯材料的零剪切粘度(zsv)与线性聚乙烯材料的zsv的比率(参看下文的antec会议录):zsv值经由上述方法在190℃下由蠕变测试获得。如上文所论述,通过常规gpc方法(等式3)测量mw(convgpc)值。线性聚乙烯的zsv与其mw(convgpc)之间的相关性基于一系列线性聚乙烯参考材料建立。zsv-mw关系的描述可以见于antec会议录:karjala等人,《低水平聚烯烃长链支化的检测(detectionoflowlevelsoflong-chainbranchinginpolyolefins)》,第66届塑料工程师协会技术年会(annualtechnicalconference-societyofplasticsengineers)(2008),887-891。1hnmr方法将储备溶液(3.26g)添加到10nmnmr管中的“0.133g聚合物样品”中。储备溶液是具有0.001mcr3+的四氯乙烷-d2(tce)和全氯乙烯的混合物(50:50,w:w)。用n2吹扫管中的溶液5分钟以降低氧气量。将加盖的样品管在室温下静置过夜以使聚合物样品膨胀。在周期涡旋混合下,将样品在110℃下溶解。样本不含可有助于不饱和度的添加剂,例如,助滑剂如芥酸酰胺。在布鲁克(bruker)avance400mhz光谱仪上在120℃下用10mm低温探针运行每个1hnmr分析。运行两个实验以得到不饱和度:对照和双重预饱和实验。对于对照实验,在lb=1hz下用指数窗函数处理数据,并且将基线从7校正到-2ppm。在对照实验中,tce的残余1h信号设定成100,并且-0.5到3ppm的积分i总用作整个聚合物的信号。聚合物中的“ch2基团数nch2”如下在等式1a计算:nch2=i总/2(等式1a)。对于双重预饱和实验,在lb=1hz下用指数窗函数处理数据,并且将基线从约6.6校正到4.5ppm。tce的残余1h信号设定成100,并且对于不饱和度(i次亚乙烯基、i三取代、i乙烯基和i亚乙烯基)的对应积分进行积分。众所周知可使用nmr光谱方法测定聚乙烯不饱和度,例如参看busico,v.等人,《大分子(macromolecules)》,2005,38,6988。次亚乙烯基、三取代、乙烯基和亚乙烯基的不饱和单元数计算如下:n次亚乙烯基=i次亚乙烯基/2(等式2a)、n三取代=i三取代(等式3a)、n乙烯基=i乙烯基/2(等式4a)、n亚乙烯基=i亚乙烯基/2(等式5a)。每1,000个碳(包含主链碳和支链碳的所有聚合物碳)的不饱和单元计算如下:n次亚乙烯基/1,000c=(n次亚乙烯基/nch2)*1,000(等式6a)、n三取代/1,000c=(n三取代/nch2)*1,000(等式7a)、n乙烯基/1,000c=(n乙烯基/nch2)*1,000(等式8a),n亚乙烯基/1,000c=(n亚乙烯基/nch2)*1,000(等式9a),对于来自tct-d2的残余质子1h信号,化学位移参考设定在6.0ppm下。对照在zg脉冲,ns=4,ds=12,swh=10,000hz,aq=1.64s,d1=14s下运行。双重预饱和实验在经改性脉冲序列下,在o1p=1.354ppm,o2p=0.960ppm,pl9=57db,pl21=70db,ns=100,ds=4,swh=10,000hz,aq=1.64s,d1=1s(其中d1是预饱和时间),d13=13s下运行。仅乙烯基含量报告于下表2中。13cnmr方法通过将约3g的含有0.025mcr(acac)3的50/50四氯乙烷-d2/邻二氯苯混合物添加到在10mmnmr管中的“0.25g聚合物样品”中,制备样品。通过用氮气吹扫管顶空从样品去除氧气。通过使用加热块和热风枪将管和其内含物加热到150℃来使样品随后溶解并且均质化。肉眼检查每个样品以确保均质性。使用布鲁克400mhz光谱仪采集所有数据。在120℃的样品温度下,使用6秒脉冲重复延迟、90度翻转角和反向门控去耦获取数据。在锁定模式下对非自旋样品进行所有测量。使样品热平衡7分钟,然后采集数据。13cnmr化学位移内部参考30.0ppm的eee三合物。c13nmr共聚单体含量:众所周知使用nmr光谱方法测定聚合物组成。astmd5017-96;jcrandall等人,“nmr和大分子(nmrandmacromolecules)”acs学术研讨会系列247;jcrandall编,《美国化学学会(am.chem.soc.)》,华盛顿哥伦比亚特区(washington,d.c.),1984,ch.9;和j.c.randall,“聚合物序列测定(polymersequencedetermination)”,学术出版社(academicpress),纽约(1977),提供了通过nmr光谱法进行聚合物分析的一般方法。分子加权共聚单体分布指数(mwcdi)来自珀里莫查公司(西班牙巴伦西亚)的gpc-ir高温色谱系统配备有精密检测器(马萨诸塞州阿姆赫斯特)、2040型2-角度激光散射检测器以及ir5红外检测器(gpc-ir)和4-毛细管粘度计(均来自珀里莫查公司)。光散射检测器的“15度角”用于计算目的。使用珀里莫查公司的仪器控制软件和数据采集界面进行数据采集。系统配备有来自安捷伦技术公司(加利福尼亚州圣克拉拉)的在线溶剂脱气装置和泵送系统。注射温度控制在150℃。使用的柱为来自聚合物实验室(英国什罗普郡)的四个20微米“mixed-a”光散射柱。溶剂是1,2,4-三氯苯。样品以“50毫升溶剂中0.1克聚合物”的浓度制备。色谱溶剂和样品制备溶剂各自含有“200ppm的丁基化羟基甲苯(bht)”。两种溶剂源都经氮气鼓泡。将乙烯类聚合物样品在160℃下温和地搅拌三小时。注射体积为“200微升”,并且流动速率为“1毫升/分钟”。用21个“窄分子量分布”的分子量在580到8,400,000g/mol范围内的聚苯乙烯标准品进行gpc柱组的校准。这些标准品布置成六个“混合液”混合物,其中单独分子量之间具有至少十倍的间隔。标准品购自聚合物实验室(英国什罗普郡)。聚苯乙烯标准物对等于或大于1,000,000g/mol的分子量以“0.025克于50毫升溶剂中”制备,并且对小于1,000,000g/mol的分子量以“0.050克于50毫升溶剂中”制备。在80℃下于轻缓搅动下溶解聚苯乙烯标准品30分钟。首先运行窄标准物混合物,并且按照“最高分子量组分”递减的次序最小化降解。使用等式1b将聚苯乙烯标准品峰值分子量转化为聚乙烯分子量(如williams和ward,《聚合物科学聚合物快报杂志(j.polym.(j.polym.sci.,polym.let.)》快报(polym.let.)》,6,621(1968)中所述):m聚乙烯=a×(m聚苯乙烯)b(等式1b),其中m是分子量,a具有约为0.40的值,并且b等于1.0。将a值调节在0.385和0.425之间(取决于特定的柱设定效率),使得nbs1475a(nist)线性聚乙烯重均分子量对应于如通过以下等式3b计算的52,000g/mol:在等式2b和3b中,rv是柱滞留体积(线性间隔),以“1点/秒”收集。ir是来自gpc仪器的测量通道的减去基线的ir检测器信号,以伏为单位,并且mpe是从等式1b测定的聚乙烯等效mw。用来自珀里莫查公司的“gpcone软件(2.013h版)”进行数据计算。使用已知短链支化(scb)频率(通过如上文所论述的13cnmr方法测量)的至少十种乙烯类聚合物标准品(聚乙烯均聚物和乙烯/辛烯共聚物;窄分子量分布和均匀的共聚单体分布)进行ir5检测器比率校准,所述标准品在均聚物(0scb/1000个总c)到约50scb/1000个总c范围内,其中总c=主链中的碳+分支中的碳。每种标准品具有如通过上文所述的gpc-lals处理方法测定为36,000g/mol到126,000g/mol的重均分子量。每种标准品具有如通过上文所述的gpc-lals处理方法测定为2.0到2.5的分子量分布(mw/mn)。scb标准品三聚合物性能示出在表1中。表1:“scb”标准品重量%共聚单体ir5面积比scb/1000个总cmwmw/mn23.10.241128.937,3002.2214.00.215217.536,0002.190.00.18090.038,4002.2035.90.270844.942,2002.185.40.19596.837,4002.168.60.204310.836,8002.2039.20.277049.0125,6002.221.10.18101.4107,0002.0914.30.216117.9103,6002.209.40.203111.8103,2002.26对于每个“scb”标准品,计算“ir5甲基通道传感器的减去基线的面积响应”与“ir5测量通道传感器的减去基线的面积响应”的“ir5面积比(或“ir5甲基通道面积/ir5测量通道面积”)”(如通过珀里莫查公司供应的标准滤光器和滤光轮:部件号ir5_fwm01包含作为gpc-ir仪器的部分)。scb频率与“ir5面积比”的线性拟合构建呈以下等式4b形式:scb/1000总c=a0+[a1×(ir5甲基通道面积/ir5测量通道面积)](等式4b),其中a0是零的“ir5面积比”下的“scb/1000个总c”截距,并且a1是“scb/1000个总c”对“ir5面积比”的斜率并且表示“scb/1000个总c”随“ir5面积比”而变的增加。将由“ir5甲基通道传感器”产生的色谱图的一系列“线性的减去基线的色谱高度”建立为柱洗脱体积的函数,以产生基线校正的色谱图(甲基通道)。将由“ir5测量通道”产生的色谱图的一系列“线性的减去基线的色谱高度”建立为柱洗脱体积的函数,以产生基线校正的色谱图(测量通道)。在样品积分界限两端的每个柱洗脱体积指数(每个等间距的指数,表示在1ml/min洗脱下每秒1个数据点)处,计算“基线校正的色谱图(甲基通道)”与“基线校正的色谱图(测量通道)”的“ir5高度比”。将“ir5高度比”乘以系数a1,并将系数a0加到此结果中,以产生样品的预测scb频率。将结果如下在等式5b中转化为共聚单体摩尔%:共聚单体摩尔百分比={scbf/[scbf+((1000-scbf*共聚单体长度)/2)]}*100(等式5b),其中“scbf”是“每1000个总c的scb”,“共聚单体的长度”对于辛烯=8,对于己烯=6,以此类推。使用williams和ward的方法(上文所述;等式1b)将每个洗脱体积指数转换成分子量值(mwi)。将“共聚单体摩尔%(y轴)”绘制为log(mwi)的函数,并且计算15,000g/mol的mwi与150,000g/mol的mwi之间的斜率(对于此计算,省略了在链端上的端基校正)。使用excel线性回归来计算15,000到150,000g/mol(并且包含端点)的mwi之间的斜率。此斜率被定义为分子加权的共聚单体分布指数(mwcdi=分子加权的共聚单体分布指数)。代表性测定mwcdi生成测量的“scb/1000个总c(=scbf)”与scb标准品的观察到的“ir5面积比”的曲线图(参见图1),并且确定截距(a0)和斜率(a1)。这里,a0=-90.246scb/1000个总c;和a1=499.32scb/1000个总c。确定样品组合物的“ir5高度比”(参见图2中所示的积分)。将该高度比(ir5高度比)乘以系数a1,并将系数a0加到该结果,以在每个洗脱体积指数下产生该实施例的预测scb频率,如上所述(a0=-90.246scb/1000总c;和a1=499.32scb/1000总c)。将scbf绘制为聚乙烯等效分子量(如使用等式1b测定,如上所讨论)的函数。参见图4(logmwi用作x轴)。通过等式5b将scbf转化为“共聚单体摩尔百分比”。如上所述,使用等式1b测定,将“共聚单体摩尔百分比”绘制为聚乙烯当量分子量的函数。参见图5(logmwi用作x轴)。线性拟合是mwi为15,000克/摩尔至mwi为150,000克/摩尔,产生“2.27摩尔%共聚单体×摩尔/克”的斜率。因此,mwcdi=2.27。使用excel线性回归来计算15,000到150,000g/mol(并且包含端点)的mwi之间的斜率。抗穿刺性在标准实验室气氛中,根据astmd5748,在测力计上在23±2℃的温度,测量耐穿刺性。仪器落镖冲击根据astmd3763测量的仪器落镖冲击是指在规定的测试条件下,用落镖撞击而刺穿材料所需的总能量。在这里,直径为12.7毫米(0.5英寸)的半球形头部落镖从66厘米的高度掉落。表2弯曲角度40°弯曲时间5s弯曲长度1mm角速度6°/秒样品80x15mm实施例以下实施例说明了本公开的特征,但无意于限制本公开的范围。使用的商用聚合物表3中列出的以下组合物包括在下面讨论的热成型单层和多层实施例中。表3*结合树脂1对应于本发明的结合层1,其具有在pct公开号wo2017/053221的表5中描述的组合物和制造方法,其通过引用整体并入本文。本发明第一组合物1-3本发明的第一组合物1-3各自包含两种乙烯-辛烯共聚物。在根据美国专利第5,977,251号(参见本专利的图2)的双串联环管反应器系统中,在如下所述的在第一反应器中存在第一催化剂系统和在如下所述的在第二反应器中存在第二催化剂系统的情况下,通过溶液聚合制备每种组合物。第一催化剂系统包括双((2-氧代基-3-(二苯并-1h-吡咯-1-基)-5-(甲基)苯基)-2-苯氧基甲基)-亚甲基-1,2-环己二基铪(iv)二甲基,由以下式(cat1)表示:原位添加到聚合反应器的cat1的金属与助催化剂1(改性的甲基铝氧烷)或助催化剂2(双(氢化动物脂烷基)甲基,四(五氟苯基)硼酸酯(1-)胺)的金属的摩尔比示于下表4中。第二催化剂系统包括齐格勒-纳塔型催化剂(cat2)。基本上根据美国专利第4,612,300号,通过以下方式制备非均匀齐格勒-纳塔型催化剂预混物:将无水氯化镁于isopare中的浆料、etalcl2于庚烷中的溶液和ti(o-ipr)4于庚烷中的溶液相继添加到一定体积的isopare中,以得到含有0.20m的镁浓度和40/12.5/3的mg/al/ti比率的组合物。用isopar-e进一步稀释此组合物的等分试样以得到浆料中500ppmti的最终浓度。在进料到聚合反应器中时并且在进入聚合反应器之前,使催化剂预混物以表4中规定的al与ti摩尔比与et3al稀释溶液接触,以得到活性催化剂。表4报告了本发明的第一组合物1-3的聚合条件。如表4所示,助催化剂1(改性甲基铝氧烷(mmao));和助催化剂2(双(氢化牛脂烷基)甲基,四(五氟苯基)硼酸酯(1-)胺、)各自用作cat1的助催化剂。测量了本发明的第一组合物1-3的其他性能,并报告在表5中。用少量(ppm)的稳定剂稳定每种聚合物组合物。表4:聚合条件(rx1=反应器1;rx2=反应器2)*溶剂=isopare表5:本发明第一组合物1-3的性质实施例1:单层膜参考下表6,根据表7提供的以下参数,在5层collin共挤出吹膜生产线上生产了数个单层膜。表6单层膜本发明第一组合物1dowlextmng2045b(lldpe)elitetmng5400b(lldpe)本发明第一组合物2dowlextmng2049b(lldpe)dowtmldpe132iversifytm2000(丙烯-乙烯共聚物)attanetm4203(uldpe)表7:单层膜厚度100μm吹胀比(bur)3.0平放宽度377mm最终宽度323mm模具类型80mm使用r145multivac热成型机,用表8中提供的以下热成型参数对所述单层膜进行热成型。表8模具尺寸包装1包装尺寸242mmx242mm.温度95℃盘最大深度110mm加热时间2秒成型时间2秒图5提供了热成型前后的单层膜的抗穿刺性。如图所示,包含本发明第一组合物1和2的单层膜显示出热成型后显著的抗穿刺性改善(例如至少10%),而对比常规单层膜通常显示出抗穿刺性降低或非常小的增长。实施例2:热成型的多层膜(流延膜挤出)表9的以下多层膜具有通过流延膜挤出制备的abcde5层结构。表9还提供了每层的厚度百分比。表9-膜样品1和2以及对比膜样品c1-c3*c1-c3代表对比膜样品1-3多层膜是在dowfreeport5层流延挤出膜生产线上生产的,表10中提供了以下特性。表10厚度7mil(177.8μm)最终宽度24.25英寸5层模切(20/20/20/20/20)使用r145multivac热成型机将表9的多层膜热成型为袋,其具有以上表8中提供的以下热成型参数。图6提供了热成型后袋的落镖性能,特别是仪器落镖性能。如图所示,包括本发明的第一组合物3的本发明样品1的仪器落镖冲击强度是对比样品c2和c3的两倍,并且是对比样品c3的大约10倍。实施例3:热成型的多层膜(吹塑膜挤出)以下表11的多层膜具有通过吹膜挤出制备的abcde5层结构。所有膜的厚度为100μm。表11-膜样品1和2以及对比膜样品c1-c3*c4-c6代表对比例4-6表11的多层膜在5层collin共挤吹膜生产线上制作,表12中提供了以下特性。表12厚度100μmbur3.0平放宽度377mm最终宽度323mm模具类型80mm使用r145multivac热成型机,使用以上表8中提供的热成型参数,将表11的多层膜热成型为袋,不同的是表11的多层膜的深度为80毫米。图7提供了热成型前后的多层膜的抗穿刺性。如图所示,包含本发明第一组合物1或2的本发明多层膜(样品3、4和6)在进行热成型后显示出改善的抗穿刺性。样品5也显示出抗穿刺性的轻微改善;但是,样品5在热成型之前已经具有很高的抗穿刺性。对比样品4(含尼龙的多层膜)、对比样品5(含尼龙和evoh的多层膜)和对比样品6(含evoh的多层膜)包括聚酰胺和/或evoh,它们通常用于热成型包装。本发明的样品(样品3-6)在利用单一材料(即仅有乙烯类聚合物)包装的同时,达到了相当的抗穿刺性和合适的落镖性能和刚度。在结合有本发明的第一组合物2和evoh的本发明样品7中,在热成型后耐穿刺性显著增加,因此表明当包含evoh和本发明第一组合物二者时有协同益处。此外,都包括本发明第一组合物2、具有0.926的密度的样品5和6显示了进一步令人惊讶的结果。如图所示,包括整体厚度的40%的本发明第一组合物2的样品5保持热成型后其抗穿刺性。然而,包括整体厚度的60%的本发明第一组合物2的样品6在热成型后其抗穿刺性显著提高。显而易见,在不脱离所附权利要求中限定的本公开的范围的情况下,修改和变化为可能的。更具体地,尽管本公开的一些方面在本文识别为优选的或特别有利的,但是预期本公开不必限于这些方面。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1