用于UV-LED固化的硫杂蒽酮衍生物光引发剂及其制备方法与应用与流程
本发明涉及一种光引发剂及其制备方法,特别是一种用于uv-led固化的具有更好的引发活性的硫杂蒽酮衍生物及其制备方法与应用。
背景技术:
光固化是指活性稀释剂、低聚物及光引发剂在光诱导之下的固化过程,一般是在成膜过程中应用,在光照下,光引发剂转变为具有活性的自由基等活性种,依此来引发活性单体与预聚物等交联聚合。
光固化技术需要用到光源,常见的有氙灯、汞弧灯、微波灯、脉冲光源以及激光光源等。它们的发射光谱从254nm变化到800nm不等,光强度也从单位mw·cm-2级别到w·cm-2。传统的光固化技术,主要使用的光源来自于紫外光(uv),常规的uv光源来自于中压或高压汞灯,但是汞灯发出的光的光谱范围较宽,因此大部分的光无法应用在光固化的过程中,而且汞灯的寿命大概在1500-2000小时之间。另外,使用汞灯有一些缺点,①能耗高:汞灯输入的能量只有大概30%可以产生紫外光,还会散发出大量热量;②长时间使用的话还会产生臭氧,对环境造成破坏;③在回收汞灯的时候,汞有一定概率会散发出去,汞有剧毒,会对环境及人体造成严重的安全问题;④紫外光会产生辐射,对人体造成影响。因此,为提高能耗、降低输出热量、保护环境等目的,寻找一种可以替代汞灯的光源有着极重要的意义。
根据中国已签署的国际公约《关于汞的水俣公约》,2021年禁止汞灯制造销售,以uv-led光源替代汞灯光源已成国际潮流,而使用led作为替代光源,有极大的帮助,目前发光二极管即led已在多个领域得到广泛的应用,例如生化医药、辐射固化及光聚合反应得到了广泛的应用。led具有几个优点是uv光源所无法达到的:①光谱宽度比较窄,宽度约为5-20nm;②光照的输出量几乎为100%,能耗低;③不产生臭氧,无紫外辐射,产生的热量低,对环境友好;④使用寿命长,运营成本低,操作简便、安全。由于以上特点,目前在工业生产领域中,使用led灯在保证工作人员的安全的同时,也能减少开支。因此,发展led光固化技术有着保证自身先进的重要意义。
一般来说,led光源的波长为395-405nm,其中395nm为uv-led固化最常用的波长。但是目前大部分的光引发剂仅在波长小于365nm范围内有着良好的吸收光性能。因此想要在led光源的照射下,光引发剂能引发各类光聚合反应:阳离子光聚合、自由基光聚合等,需要在光引发剂中引入官能团,增加其刚性结构与共轭程度,使红外光谱红移,在较高的波长下也能很好的吸收光照,引发光聚合反应。
与紫外光对比起来,可见光的能量较弱,发射谱带比较窄,难以使裂解型光引发剂裂解,所以led光引发剂通常是抽氢型光引发剂。硫杂蒽酮类衍生物是一种抽氢型光引发剂,由于其紫外吸收光谱波长高,引发效率高等缘故,目前在led光引发剂中研究较为热门,但是它必须和胺助引发剂配合使用,硫杂蒽酮吸收光能后,经激发三线态与助引发剂作用形成基激复合物,经电子转移和夺氢反应,产生具有较高引发活性的胺烷基自由基引发聚合。但是,胺类化合物具有毒性和致癌性,而且由其引发所得的固化膜易发生黄变,制约了其在食品和药物包装等方面的应用。
技术实现要素:
为了克服上述缺陷,本发明要解决的技术问题是:针对硫杂蒽酮的特性以及当前led光引发剂的趋势,提供一种制备成本低,吸收波长在可见光范围内的用于uv-led固化的硫杂蒽酮衍生物光引发剂及其制备方法,代替硫杂蒽酮直接作为光引发剂使用,不需要胺助引发剂,以减少由胺助引发剂引起的黄变和毒性,避免胺类化合物使用带来的毒性和致癌性。
本发明另一目的在于提供所述的uv-led固化的硫杂蒽酮衍生物类光引发剂在涂料中的应用。
本发明光引发剂硫杂蒽酮衍生物在硫杂蒽酮的特殊结构上对分子结构进行修饰,使硫杂蒽酮衍生物红移,使吸收波长达到led发射光波长区间,因此可以代替硫杂蒽酮直接作为光引发剂使用,不需要胺助引发剂,以减少由胺助引发剂引起的黄变和毒性,而且其制备成本低,操作简单,具有较高的引发活性;
光引发剂硫杂蒽酮衍生物中的硫杂蒽酮结构式:
本发明目的通过如下技术方案实现:
用于uv-led固化的硫杂蒽酮衍生物类光引发剂的制备方法,包括如下步骤:
1)缩合成环反应:在0-5℃的水浴环境下,容器中加入浓硫酸后,在搅拌下分批加入硫代水杨酸,待溶液搅拌混合均匀后;20-30min内将苯酚衍生物滴加到反应器中,添加完毕后保持0-5℃搅拌1-2h;观察到反应溶液颜色变深,产物转移到70-80℃的油浴环境中,继续搅拌,反应4-6h;溶液颜色为暗红色后,静置过夜;将反应器中的溶液边搅拌边倒入冷冻至0-5℃的蒸馏水,直至出现沉淀,抽滤,水洗,得到固体粗产品;
2)将氢氧化钠固体加到蒸馏水中配置氢氧化钠溶液;将吸干后的粗产品溶解于氢氧化钠溶液中,过滤,得到橙红色滤饼以及滤液;将滤液酸化至ph为6-7,得到橙黄色浑浊溶液,静置溶液待沉淀凝聚与母液分层后,将沉淀部分离心,得沉淀固体,干燥,与滤饼合并,得到硫杂蒽酮衍生物类光引发剂;
所述苯酚衍生物为邻甲酚、间甲酚、对甲酚、2,4-二甲基苯酚和邻苯二酚、间苯二酚和对苯二酚的一种或多种;
为进一步实现本发明目的,优选地,步骤1)的反应体系中硫代水杨酸与浓硫酸的摩尔比为1:15-40,硫代水杨酸与苯酚衍生物的摩尔比为1:3-6。
优选地,步骤2)所述的氢氧化钠溶液由氢氧化钠固体与蒸馏水按质量质量比1:12.5-13.0配制。
优选地,步骤2)所述的滤液酸化的温度为20-30℃。
优选地,步骤1)所述的浓硫酸的体积浓度为98%。
优选地,步骤1)所述的浓硫酸与蒸馏水的质量比为1:5-12;所述蒸馏水为二次蒸馏水。
优选地,步骤1)所述的抽滤是在出现沉淀再搅拌5-15min后进行;所述的水洗次数为多次。
优选地,步骤2)所述的吸干为真空吸干;所述的滤液酸化是通过加入盐酸溶液进行;步骤2)所述的将沉淀部分离心是静置溶液待沉淀凝聚与母液分层后用胶头滴管吸出母液部分,并将沉淀部分移入离心管中,放入离心机离心10-20分钟后倒掉溶液并刮出沉淀固体;步骤2)所述的干燥是置于60-70℃烘箱干燥。
一种用于uv-led固化的硫杂蒽酮衍生物类光引发剂,由上述制备方法制得;所述uv-led固化的硫杂蒽酮衍生物类光引发剂的吸收波长为390-405nm。
所述的uv-led固化的硫杂蒽酮衍生物类光引发剂在涂料中的应用:将20-26%质量的bayer的聚氨酯丙烯酸树脂uaxp2513、20-28%质量的液体环氧丙烯酸树脂、40-56%的活性稀释剂与4-6%质量的硫杂蒽酮衍生物类光引发剂混合均匀得涂料;所述的硫杂蒽酮衍生物类光引发剂预先溶解在活性稀释剂中;所述的活性稀释剂由hdda与tmpta按照质量比1:1-1.5混合而成;
所述的环氧丙烯酸树脂为双酚a型环氧树脂e55、e51、e44和e31与丙烯酸的缩聚产物一种或多种。
与现有技术相比,本发明的优点和有益效果为:
(1)本发明光引发剂具有低挥发性、无气味、低黄变的特点,引发活性与小分子的硫杂蒽酮相当,针对硫杂蒽酮迁移性强、有毒的缺点,有效的填补硫杂葱酮在某些应用领域应用的空白,随着人们环保意识的增强,未来也将成为硫杂葱酮的替代品。
(2)本发明修饰了光引发剂的分子结构,引发剂不需再额外添加活性胺即可引发,属自引发夺氢型光引发剂,生产上进一步降低成本。
(3)本发明的化合物是一种低挥发性、低黄变、合成简单、价格便宜的硫杂蒽酮替代品。
附图说明
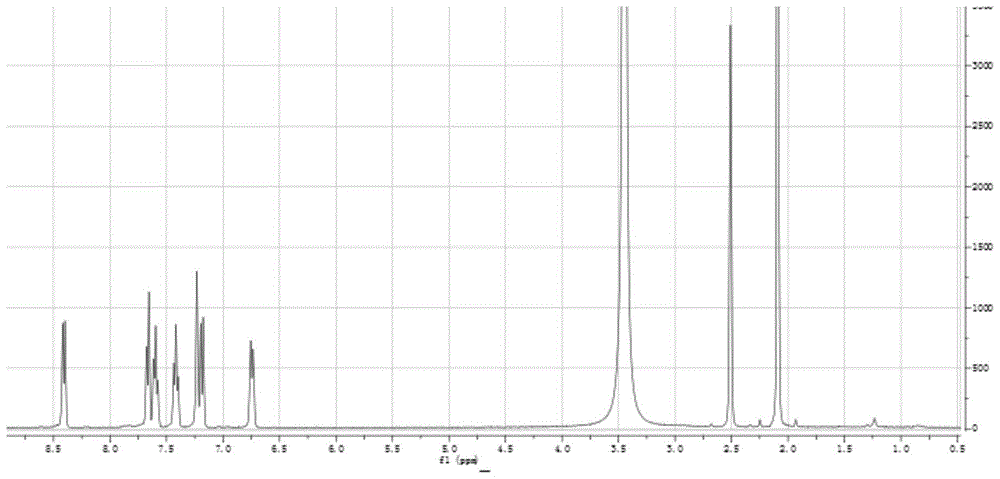
图1为实施例1中硫杂蒽酮衍生物的核磁氢谱。
图2为实施例1中硫杂蒽酮衍生物的红外图谱。
图3位实施例中1中硫杂蒽酮衍生物在dmso中的紫外吸收图谱。
具体实施方式
为更好地理解本发明,下面结合实施例进一步说明本发明,但实施例不构成对本发明保护范围的限制。
实施例中所用的酸为直接为商业购买的酸,没有进行任何的稀释。
实施例1:
本实施例一种硫杂蒽酮衍生物的反应式:
(1)缩合成环反应:在0℃的水浴环境下,加入15ml体积浓度为98%浓硫酸后,在转子搅拌器的搅拌下分批加入1.54g硫代水杨酸,待溶液搅拌混合均匀后。称取5.64g苯酚,20min滴入到反应器中,添加完毕后保持0℃搅拌1h。观察到溶液颜色变深,于是转移到70℃的油浴环境中,继续搅拌,反应5h。静置过夜。将反应器中的溶液边搅拌边倒入0℃的50ml二次蒸馏水中,直至出现沉淀,搅拌5min后抽滤,多次水洗得到橘红色固体粗产品。
(2)将粗产品真空吸干后,称量取32g氢氧化钠固体,搅拌加到400ml蒸馏水中配置氢氧化钠溶液。将粗产品溶解于氢氧化钠溶液中,过滤,得到橙红色滤饼以及滤液。以2m盐酸溶液,在25℃下将滤液酸化至ph=6,得到橙黄色浑浊溶液,静置溶液待沉淀凝聚与母液分层后,用胶头滴管吸出母液部分,并将沉淀部分移入离心管中,放入离心机离心10分钟后倒掉溶液并刮出沉淀固体,沉淀固体置于70℃烘箱干燥,与前面滤饼所得固体合并,得到橙红色固体产物tx-1。
所得产物性能采用以下方法检测:核磁氢谱测定:使用氘代二甲基亚砜(dmso-d6)作为溶剂,5ml溶剂溶解5mg产物进行核磁氢谱测试;红外图谱测定:使用光谱纯的溴化钾作为测试背景通道,并取用量为溴化钾1/200的样品与溴化钾混合研磨至200目以下,干燥后取2mg压片,放置入红外光谱仪中进行测试;根据gb/t6739-2006测定涂膜硬度;根据gb/t23983-2009测定涂层的耐黄变性;
利用紫外可见光谱仪测定光引发剂溶液中的吸收波长;光引发剂挥发性测定:将1mg样品置于恒温箱24h并保持85℃,对比放置前后质量差,并计算质量损失率;涂膜中引发剂迁移率测量:称取的1g固化膜样品于的锥形瓶并用20ml乙腈溶解,采用超声萃取30min,将萃取液蒸干溶液,再加入2ml乙腈溶解萃取所得固体,并且经过0.45μm滤膜过滤,收集所得滤液采用气相色谱质谱法gc-ms分析。
测试结果:
1)图1为硫杂蒽酮衍生物的核磁,6.77-8.47ppm处有七个特征吸收峰,代表了两个苯环上的七个氢原子;并且谱图在3.48ppm左右均有一个氢原子峰,可以判定这个为羟基上氢原子的特征吸收峰。图2红外图谱所示,3619cm-1处的吸收峰代表的是羟基;1584cm-1处的吸收峰代表的是羰基;1468cm-1处的吸收峰代表的是苯环上的碳碳双键;1277cm-1处的吸收峰代表的是碳氧键;737cm-1处的吸收峰代表的是碳硫键。通过上述分析,可从该红外光谱看出产物有羰基、羟基、碳碳双键、碳氧键及碳硫键的存在,符合目标产物的官能团。结合图1的核磁共振氢谱图,证实该产物与目标产物的结构基本相符。
2)室温条件下将tx-1配制得1×10-4mol·l-1二甲基亚砜(dmso)溶液,并以空白二甲基压砜溶液作为参比,得图3。可发现tx-1的稀溶液最大吸收波长=404nm。
3)将20%质量的uaxp2513和25%质量的e55环氧丙烯酸树脂与5%质量的光引发剂橙红色固体产物tx-1混合均匀,得涂料。光引发剂预先溶解在活性稀释剂中;活性稀释剂由占原料总质量20%的hdda与30%的tmpta组成。将所得涂料均匀涂在干净的玻璃板上,将玻璃板放在光固化机中辐照。实验性能结果见表1。
表1不同光引发剂的性能对比
经过一系列的测试验证以及表1的光引发剂性能对比的结果可以看出,本发明通过硫代水杨酸一步法与苯酚反应,在苯环引入羟基作为供电子基团,形成一种推拉电子结构的硫杂蒽酮衍生物,图3的吸收图谱证明该引发剂的激发波长的确在led灯的光波长处。
与商用的itx相比,本实施例无需与胺助引发剂(edab)作用形成基激复合物,可直接产生具有较高引发活性碎片从而引发聚合,其引发速率相当,降低引发剂的黄变。
进一步地与edb相比,本实施例tx-1引发剂转化率更高,从而使得涂膜交联程度更高,硬度更高,同时固体挥发性明显比前两者更低。而考虑到本实施例tx-1本身是用能量较低的405nm波长进行固化,所以固化时间偏长,但是并不影响tx-1在综合性能上与商用的光引发剂相比,可以对比其挥发性以及在涂膜中迁移率的数据,可见tx-1具有低迁移率,低挥发性等优点的突出。
实施例2
(1)缩合成环反应:在5℃的水浴环境下,加入20ml98%浓硫酸后,在转子搅拌器的搅拌下分批加入1.6g硫代水杨酸,待溶液搅拌混合均匀后。尽量快的用电子分析天平量取3.36g对甲酚,并分批加入到反应器中,添加完毕后保持0℃搅拌2h。观察到溶液颜色变深,于是转移到70℃的油浴环境中,继续搅拌,反应4h。静置过夜。将反应器中的溶液边搅拌边倒入50ml的0℃冷水中,直至出现沉淀,搅拌10min后抽滤,多次水洗得到橘红色固体粗产品。
(2)将粗产品真空吸干后,称量取28g氢氧化钠固体,搅拌加到360ml蒸馏水中配置氢氧化钠溶液。将粗产品溶解于其中,过滤,得到橙黄色滤饼以及滤液。以2m盐酸溶液,在30℃下将滤液酸化至ph=6.5,得到橙黄色浑浊溶液,静置溶液待沉淀凝聚与母液分层后,用胶头滴管吸出母液部分,并将沉淀部分移入离心管中,放入离心机离心15分钟后倒掉溶液并刮出沉淀固体,置于60℃烘箱干燥,与前面滤饼所得固体合并得到橙黄色固体产物tx-2。
测定结果:
1)核磁氢谱与红外光谱证明,成功地合成设计的分子结构。
2)室温条件下将tx-2配制得1×10-4mol·l-1二甲基亚砜(dmso)溶液,并以空白二甲基压砜溶液作为参比。溶液最大吸收波长max2=400nm。属于可见光范围内。
3)将24%质量的uaxp2513与22%质量的e44环氧丙烯酸树脂分别与4%质量的光引发剂(预先溶解在活性稀释剂25%质量hdda与25%质量tmpta中)混合均匀,将所得涂料均匀涂在干净的玻璃板上,将玻璃板放在光固化机中辐照。实验性能结果见表2。
表2不同光引发剂的光固化效果
实施例3:
(1)缩合成环反应:在2℃的水浴环境下,加入18ml98%浓硫酸后,在转子搅拌器的搅拌下分批加入1.8g硫代水杨酸,待溶液搅拌混合均匀后。尽量快的用电子分析天平量取7.57g邻苯二酚,并分批加入到反应器中,添加完毕后保持2℃搅拌1h。观察到溶液颜色变深,于是转移到70℃的油浴环境中,继续搅拌,反应5.5h。静置过夜。将反应器中的溶液边搅拌边倒入2℃的50ml冷水中,直至出现沉淀,搅拌15min后抽滤,多次水洗得到橘黄色粗产品。
(2)将粗产品真空吸干后,称量取30g氢氧化钠固体,搅拌加到390ml蒸馏水中配置氢氧化钠溶液。将粗产品溶解于其中,过滤,得到橙黄色滤饼以及滤液。以2m盐酸溶液,在25℃下将滤液酸化至ph=7,得到橙黄色浑浊溶液,静置溶液待沉淀凝聚与母液分层后,用胶头滴管吸出母液部分,并将沉淀部分移入离心管中,放入离心机离心10分钟后倒掉溶液并刮出沉淀固体,置于65℃烘箱干燥,与前面滤饼所得固体合并得到橙红色固体产物tx-3。
测试结果:
1)核磁氢谱与红外光谱证明,成功地合成设计的分子结构。
2)室温条件下将tx-3配制得1×10-4mol·l-1二甲基亚砜(dmso)溶液,并以空白二甲基压砜溶液作为参比。溶液最大吸收波长max3=403nm,属于可见光范围内。
3)将24%质量的四官能团聚氨酯丙烯酸酯与20%质量的环氧丙烯酸树脂分别与6%质量的光引发剂(预先溶解在活性稀释剂30%质量hdda与20%质量tmpta中)混合均匀,将所得涂料均匀涂在干净的玻璃板上,将玻璃板放在光固化机中辐照。实验性能结果见表3。
表3不同光引发剂的光固化效果
需要说明的是,本发明不受上述实施例的限制,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明保护范围内;本发明要求保护范围由权利要求书界定。
- 还没有人留言评论。精彩留言会获得点赞!