一种以邻苯二酚为原料全合成小檗红碱的方法与流程
本发明涉及一种药物合成方法,特别是涉及一种以邻苯二酚为原料全合成小檗红碱的方法。
背景技术:
小檗红碱(berberrubine)又称9-脱甲基小檗碱,9-脱甲基黄连素等,因其红色而得名。分子式为c19h16no4cl,分子质量为321.33u,是高共轭电中性醌式结构,暗红色针状晶体,熔点280℃-282℃,碘化铋钾反应和亚甲二氧基反应阳性。与小檗碱(berberine)一样,是黄连等中药材的有效成分之一,属于原小檗碱类化合物,是一类异喹啉生物碱。已有研究和临床应用结果表明,原小檗碱类化合物具有抗肿瘤、降血糖、抗炎、降血脂、抗病原微生物、抗阿尔茨海默病、抗老年痴呆、抗心律失常等广泛的药理活性。由于黄连等中药材中小檗红碱的含量较低,同时,又是共同疗效,一直以来,研究多集中于药物的性质和结构鉴定、药理药效等,对各种成分的分离鉴定研究较少,没有单独使用也没有被重视。
小檗红碱最先是从berberisevalgarisl分离得到的一种生物碱,从黄连中也能分离得到该化合物。小檗碱加热分解脱甲基可得到小檗红碱,研究表明,小檗碱体内代谢主要产物之一是小檗红碱。目前,化学合成小檗红碱的方法就是将小檗碱脱甲基得到小檗红碱,脱甲基的方法有加热热解法、吡啶盐酸盐熔融法、尿素法、氯化铝催化法、先酯化后水解法,但是,不难发现,目前,小檗红碱的制备都是以小檗碱为原料,这不仅消耗了宝贵的小檗碱药物,而且增加了药物成本。
小檗碱本身也是抗炎抗菌有效药物,对其进行脱甲基制备小檗红碱,一方面,消耗了小檗红碱的产量,另一方面,在小檗碱的基础上制备小檗红碱,生产成本继续增加,少量研究使用没有太大影响,大量临床应用必然会造成成本增加,同时受到小檗碱的产量的限制,不利于小檗红碱药物的扩大使用,开展小檗红碱的全合成研究和开发,具有创新性。
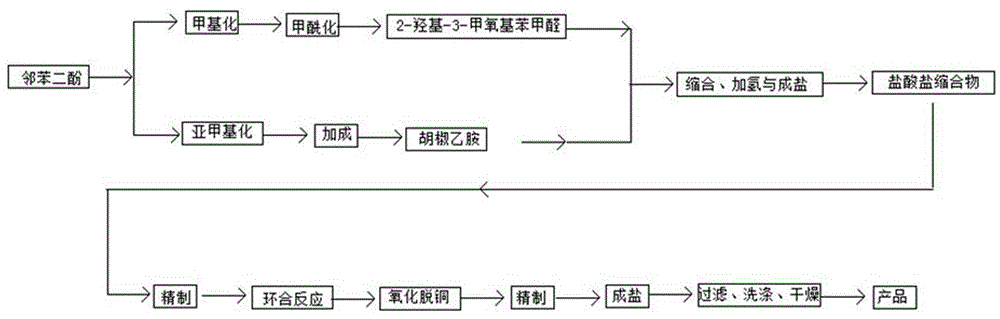
以邻苯二酚为原料,化学全合成小檗红碱,采用的原料为大宗有机原料邻苯二酚,通过邻苯二酚与2-氯甲烷选择性甲基化反应与甲酰化反应得到2-羟基-3-甲氧基苯甲醛;通过邻苯二酚与2-氯甲烷进行亚甲基化反应与一步法催化加成反应得到胡椒乙胺,实现了胡椒环一步法生产胡椒乙胺,缩短了胡椒乙胺合成步骤,克服了有毒氰化物的使用,工艺绿色化,可持续化。采用“一锅法”缩合加氢成盐,工艺过程中溶剂回收循环利用,节约了时间,节约了能源,降低了成本,实现了工业化生产小檗红碱,对最大效果地发挥和利用小檗红碱抗病治病,在当前条件下具有重要意义。
技术实现要素:
本发明的目的在于提供一种以邻苯二酚为原料全合成小檗红碱的方法,本发明通过选择性甲基化与甲酰化得到2-羟基-3-甲氧基苯甲醛;通过亚甲基化反应得到胡椒环后,采用一步法催化加成反应合成胡椒乙胺,缩短了胡椒乙胺的合成步骤,避免了使用有毒氰化物,工艺绿色化,可持续化;缩合加氢与成盐反应采用“一锅法”,节约了时间,节约了能源,降低了成本。
本发明的目的是通过以下技术方案实现的:
一种以邻苯二酚为原料全合成小檗红碱的方法,所述方法包括以下制备过程:
第一步,以邻苯二酚为原料,其纯度(w%)99,二甲基亚砜为溶剂,碳酸钾为缚酸剂,与二氯甲烷在130℃条件下,进行亚甲基化反应得到胡椒环;第二步,以乙酸乙酯为溶剂,胡椒环与2-氯乙胺在65℃以及催化剂条件下,一步法加成反应得到胡椒乙胺;第三步,以无水乙醇为溶剂,聚乙二醇和氢氧化钠为催化剂,邻苯二酚与二氯甲烷在60℃条件下,甲基化反应得到邻甲氧基苯酚;第四步,以乙腈为溶剂,三乙胺为缚酸剂,氯化镁为催化剂,邻甲氧基苯酚与多聚甲醛在65℃条件下,选择性甲酰化反应得到2-羟基-3-甲氧基苯甲醛;第五步,2-羟基-3-甲氧基苯甲醛与胡椒乙胺分别加入反应釜内,真空条件下,85℃,缩合反应25min.,保温30min.;降温后,继续投入镍基催化剂和溶剂,开启加压,加氢,升温至115℃,反应50min.继续保温20min.,产物降温至75℃,开启减压,蒸出部分溶剂;反应生成物加入盐酸,降温结晶,过滤得到结晶物;第六步,将盐酸盐缩合物晶体,加入热水中精制,除去杂质;第七步,将结晶物与乙二醛,在醋酸醋酐溶剂中,加入铜基催化剂,120℃条件下,保温反应2h,进行环合反应;反应液降温至80℃,减压蒸出部分溶剂;第八步,反应液降温至室温,向反应产物中加入盐酸和氧化剂,进行精制反应;第九步,向反应液中加入氨水,升温至80℃,并加入活性炭,进行精制反应,过滤得到反应液;第十步,向反应液中加入盐酸,降温结晶,过滤得到产物晶体初品;第十一步,用乙醇洗涤,干燥,得到产品小檗红碱。
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述制备得到的胡椒乙胺与2-羟基-3-甲氧基苯甲醛经过缩合反应得到席夫碱,继续加氢后,加入盐酸成盐反应得到盐酸盐缩合物。
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述将盐酸盐缩合物与乙二醛进行催化环合反应得到小檗红碱反应液。
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述将反应液加入盐酸和双氧水,脱去催化剂氯化铜。
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述将脱铜精制液继续加入氨水和活性炭,进行精制反应;
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述将过滤后得到的精制液,加入盐酸,降温结晶,过滤得到粗产品。
所述的一种以邻苯二酚为原料全合成小檗红碱的方法,所述过滤得到的粗产品,经乙醇洗涤后,过滤、干燥得到产品小檗红碱。
本发明的优点与效果是:
本发明工艺过程实现了工业化全合成生产小檗红碱,采用的原料为大宗有机原料邻苯二酚,原料易得,价格便宜;通过选择性甲基化与甲酰化得到2-羟基-3-甲氧基苯甲醛;通过亚甲基化反应得到胡椒环后,采用一步法催化加成反应合成胡椒乙胺,缩短了胡椒乙胺的合成步骤,避免了使用有毒氰化物,工艺绿色化,可持续化;缩合加氢与成盐反应采用“一锅法”,节约了时间,节约了能源,降低了成本。工业化全合成生产小檗红碱,开创了小檗红碱药物的化学合成,并且,可规模化生产小檗红碱,满足了小檗红碱在当前抗肿瘤、抗血压、抗心律、降血糖、治疗阿尔茨海默病等的临床与研究需要,为减低病人痛苦提供了有效药物的同时,经济效益和社会效益显著。
1、以邻苯二酚为原料,属大宗有机化工原料,原料来源广,易得,为大规模生产小檗红碱提供了保证;
2、采用选择性甲基化与甲酰化技术得到2-羟基-3-甲氧基苯甲醛;
3、采用一步法催化加成反应得到胡椒乙胺,即缩短了工艺过程,又避免了使用有毒氰化物,工艺绿色化,可持续化;
4、采用“一锅法”进行缩合加氢与成盐反应,节约了时间,节约了能源,降低了成本;
5、工艺技术实现了工业化全合成生产小檗红碱;
6、工艺过程中对溶剂回收,提纯后,循环利用;
7、以邻苯二酚为原料生产小檗红碱,不消耗小檗碱产品。
8、对缩合加氢结晶物进行精制反应,保证了环合反应的质量。
附图说明
图1为本发明工艺流程方框图;
图2为中间体红外谱;
图3为中间体核磁共振谱图;
图4为红外谱图;
图5为核磁共振谱图。
具体实施方式
下面结合实施例对本发明进行详细说明。
本发明以邻苯二酚为原料,一方面,邻苯二酚与2-氯甲烷选择性甲基化反应得到邻甲氧基苯酚,继续选择性甲酰化反应得到2-羟基-3-甲氧基苯甲醛;另一方面,邻苯二酚与2-氯甲烷亚甲基化反应得到胡椒环,继续与2-氯乙胺催化加成反应得到胡椒乙胺;将胡椒乙胺与2-羟基-3-甲氧基苯甲醛在镍基催化剂条件下“一锅法”缩合加氢反应,反应产物加盐酸,冷却结晶物,过滤得到盐酸盐缩合物;将盐酸盐缩合物精制后,继续与乙二醛,在铜基催化剂条件下,进行环合反应,反应产物经过精制过程后,加盐酸,冷却结晶,过滤洗涤得到产物小檗红碱。
本发明以邻苯二酚为原料,其纯度(w%)99,二甲基亚砜为溶剂,碳酸钾为缚酸剂,与二氯甲烷在130℃条件下,进行亚甲基化反应得到胡椒环;以乙酸乙酯为溶剂,胡椒环与2-氯乙胺在65℃以及催化剂条件下,一步法加成反应得到胡椒乙胺;以无水乙醇为溶剂,聚乙二醇和氢氧化钠为催化剂,邻苯二酚与二氯甲烷在60℃条件下,甲基化反应得到邻甲氧基苯酚;以乙腈为溶剂,三乙胺为缚酸剂,氯化镁为催化剂,邻甲氧基苯酚与多聚甲醛在65℃条件下,选择性甲酰化反应得到2-羟基-3-甲氧基苯甲醛;2-羟基-3-甲氧基苯甲醛与胡椒乙胺分别加入反应釜内,真空条件下,85℃,缩合反应25min.,保温30min.;降温后,继续投入镍基催化剂和溶剂,开启加压,加氢,升温至115℃,反应50min.继续保温20min.,产物降温至75℃,开启减压,蒸出部分溶剂;反应生成物加入盐酸,降温结晶,过滤得到结晶物;将盐酸盐缩合物晶体,加入热水中精制,除去杂质;将结晶物与乙二醛,在醋酸醋酐溶剂中,加入铜基催化剂,120℃条件下,保温反应2h,进行环合反应;反应液降温至80℃,减压蒸出部分溶剂;反应液降温至室温,向反应产物中加入盐酸和氧化剂,进行精制反应;向反应液中加入氨水,升温至80℃,并加入活性炭,进行精制反应,过滤得到反应液;向反应液中加入盐酸,降温结晶,过滤得到产物晶体初品;用乙醇洗涤,干燥,得到产品小檗红碱。
1.全合成工艺过程
(1)全合成工艺
(2)“一锅法”缩合加氢原理
席夫碱催化加氢反应中镍基催化剂,反应在高压反应釜中进行,纳米颗粒的镍金属催化剂的表面积大大增加,极大的表面积带来的是很高的催化活性,对氢气的强吸附性以及热稳定性。
(3)缩合与环合反应原理
2.本发明的基本方案
第一步,以邻苯二酚为原料,与二氯甲烷进行亚甲基化反应得到胡椒环;第二步,以乙酸乙酯为溶剂,胡椒环与2-氯乙胺在65℃以及催化剂条件下,一步法加成反应得到胡椒乙胺;第三步,以无水乙醇为溶剂,聚乙二醇和氢氧化钠为催化剂,邻苯二酚与二氯甲烷在60℃条件下,甲基化反应得到邻甲氧基苯酚;第四步,以乙腈为溶剂,三乙胺为缚酸剂,氯化镁为催化剂,邻甲氧基苯酚与多聚甲醛在65℃条件下,选择性甲酰化反应得到2-羟基-3-甲氧基苯甲醛;第五步,2-羟基-3-甲氧基苯甲醛与胡椒乙胺分别加入反应釜内,真空条件下,85℃,缩合反应25min.,保温30min.;降温后,继续投入镍基催化剂和溶剂,开启加压,加氢,升温至115℃,反应50min.继续保温20min.,产物降温至75℃,开启减压,蒸出部分溶剂;反应生成物加入盐酸,降温结晶,过滤得到结晶物;第六步,将盐酸盐缩合物晶体,加入热水中精制,除去杂质;第七步,将结晶物与乙二醛,在醋酸醋酐溶剂中,加入铜基催化剂,120℃条件下,保温反应2h,进行环合反应;反应液降温至80℃,减压蒸出部分溶剂;第八步,反应液降温至室温,向反应产物中加入盐酸和氧化剂,进行精制反应;第九步,向反应液中加入氨水,升温至80℃,并加入活性炭,进行精制反应,过滤得到反应液;第十步,向反应液中加入盐酸,降温结晶,过滤得到产物晶体初品;第十一步,用乙醇洗涤,干燥,得到产品小檗红碱。
3.本发明的技术方法
本发明以邻苯二酚为原料,采用亚甲基化反应得到胡椒环,采用催化胺化一步法反应得到胡椒乙胺;以邻苯二酚为原料,采用选择性甲基化反应得到邻甲氧基苯酚,采用选择性催化甲酰化反应得到2-羟基-3-甲氧基苯甲醛;将反应得到胡椒乙胺与2-羟基-3-甲氧基苯甲醛采用“一锅法”进行缩合脱水与加压催化加氢,加盐酸成盐制得中间体。中间体产物继续与乙二醛,在铜基催化剂条件下,进行环合反应制得初产品,经过氨铜络合以及活性炭精制后,加入盐酸,降温结晶,经过洗涤、干燥后得到小檗红碱。
4.本发明具体的实施方案
(1)按照摩尔比称取邻苯二酚与2-氯甲烷,以二甲基亚砜为溶剂,投入到500ml三口烧瓶中,加入缚酸剂(碱),130℃条件下反应6h,减压蒸馏回收部分2-氯甲烷,收集130-135℃产物,得到目标产物胡椒环。
(2)以乙酸乙酯为溶剂,按照摩尔比称取胡椒环与2-氯乙胺,投入到500ml三口烧瓶中,加入催化剂,滴加2-氯乙胺,搅拌反应8h,冷却后,减压整出乙酸乙酯,-10℃结晶,真空过滤,乙醇重结晶得到胡椒乙胺。
(3)按照质量比1:4,称取邻苯二酚和氢氧化钠,投入500ml三口烧瓶中,加入催化剂(醇类),以甲醇为溶剂,升温搅拌至固体溶解,65℃条件下,滴加2-氯甲烷,保温反应3h,过滤,减压浓缩回收溶剂,用环己烷萃取,蒸出甲醇得到产品邻甲氧基苯酚。
(4)以乙腈为溶剂,称取18g邻甲氧基苯酚,倒入500ml三口烧瓶中,加入催化剂(路易斯酸),升温搅拌至65℃,滴加三乙胺,加入多聚甲醛,慢慢升温至回流,反应1h,降至室温加入盐酸调节ph值,得到2-羟基-3-甲氧基苯甲醛。
(5)称取一定量(摩尔比)的胡椒乙胺与2-羟基-3-甲氧基苯甲醛放入250ml高压釜中,搅拌升温至70℃,减压蒸馏(压力0.095mp),继续升温至95℃,无水蒸出即为反应终点,保温30min,席夫碱制备完成。
向高压釜加入镍基催化剂0.25g,通氮气排出釜内空气,在釜内氢压3mp的情况下,升温至70℃,升压至4mp,温度升至115℃,关闭氢气阀门,直至压力表示数不在下降为止,补压至4mp,在4mp下保温、保压50min,降温静止,得到缩合氢化液。
缩合氢化液降温至75℃,,开始常温蒸溶剂(回收循环利用),蒸出适量溶剂。
向残余液中加入盐酸,降温结晶,过滤得到盐酸盐缩合物。上述过程始终在反应釜内进行,简称“一锅法”缩合加氢成盐。
(6)将得到的盐酸盐缩合物100g,投入到70-90℃的热水中,分层精制,取中间层重结晶,得到精制的盐酸盐缩合物。
(7)用量筒量取冰乙酸(分析纯)、醋酐(工业品),电子天平称取无水氯化铜(工业品)加入到500ml三口烧瓶内,插入搅拌浆,转速控制150r/min;打开恒温水浴锅,升温至85℃,冷凝回流,滴加预热好的乙二醛(含量64~66%),继续升温至115℃,加入盐酸盐缩合物,控制转速在250r/min,在110℃~120℃保温反应2小时。环合反应结束。
(8)将环合反应后的三口烧瓶连接上蒸馏装置,加入转子,调节水浴锅温度在80℃,转速控制在200r/min,反应在一小时后,水浴锅温度调节至55℃,蒸馏装置连接上真空泵,压力控制在0.3个大气压,减压蒸溶剂;两个小时反应完成,收集溶剂液体,蒸溶剂结束。
(9)蒸溶剂结束后,向三口烧瓶中加入若干水,调节温度在80℃,转速控制在200r/min,搅拌40分钟,搅拌结束后,加水至适量,降温至常温以下准备脱铜。用量筒量取盐酸(含量37%)加入三口烧瓶中。关闭恒温水浴锅,降温,温度降至常温以下,打开搅拌器,转速控制在200r/min,开始缓慢滴加已备好的双氧水(纯量4.5g,含量30%,密度1.11g/cm),温度降至常温以下过滤,用ph=2的酸水洗涤滤渣。
(10)将产物移至5l三口烧瓶中加大量蒸馏水,转速控制在200r/min,搅拌30min,加入氨水,搅拌40min,升温至85℃,加入活性炭,在90℃保温40min,过滤,用热水洗涤滤渣,将滤液移至5l三口烧瓶中,转速控制在200r/min,搅拌30min,加入氨水,搅拌40min,升温至90℃,加入活性炭,在90℃保温40min,过滤,用热水洗涤滤渣,
(11)在5l三口烧瓶中加入浓盐酸适量(含量37%),将滤液转移至三口烧瓶中。搅拌降温,结晶至常温以下为止,过滤,干燥。
以2-羟基-3-甲氧基苯甲醛为原料,经缩合加氢成盐的盐酸盐缩合物红外谱图如图2所示,根据其分子结构,由图2可知,在1,606cm-1,1504cm-1,1489cm-1,1447cm-1处的伸缩振动;结合990cm-1,812.01cm-1,771.9cm-1以及705.23cm-1处的c-h弯曲振动,可以判断为芳烃(苯环)及取代基结构;在1040.99cm-1处为c-o-c伸缩振动,结合921.99cm-1及934.8cm-1处c-h的强振动,归属于次甲基二氧连苯环(胡椒环)结构,而在1247.12cm-1处为ar-o的伸缩振动,结合2820cm-1处的振动峰为-och3;在3434cm-1处较宽的峰,结合1298.3cm-1处c-o的伸缩振动以及765.23cm-1处-oh的面外弯曲,可以归属于ar-oh结构;在2051.72cm-1处的弱的振动吸收,结合1357cm-1处c-n弱的伸缩振动吸收,1619.3cm-1与1606.08cm-1处n-h变形振动以及在900~650cm-1面外弯曲振动,可归属于-hcn亚胺盐结构。
中间体核磁共振谱图3
由盐酸盐缩合物1h-nmr核磁谱图3可知,δppm:6.88、6.87、6.85、6.73和6.71(1h,苯环),5.99(2h,-o-c-o-),2.86和2.83(2h,-ch2-),3.85和3.51(3h,-och3),2.52(五重峰,氘代dmso溶剂峰),3.51(样品含水峰)。
由图4可知,以2-羟基-3-甲氧基苯甲醛为原料,经缩合加氢成盐的盐酸盐缩合物,继而与乙二醛环合得到产物小檗红碱,由红外谱图4可知,在1502.61cm-1,1479.37cm-1处的伸缩振动;结合873.1cm-1处的c-h弯曲振动,可以判断为芳烃(苯环)及取代基结构;在1033.91cm-1处为c-o-c伸缩振动,结合930.54cm-1处c-h的强振动,归属于次甲基二氧连苯环(胡椒环)结构,而在1231.60cm-1处为ar-o的伸缩振动,结合2820cm-1处的振动峰为-och3;在3429.20cm-1处较宽的峰,结合1287.05cm-1处c-o的伸缩振动以及765.23cm-1处-oh的面外弯曲,可以归属于ar-oh结构;在2051.72cm-1处的弱的振动吸收,结合1352.33cm-1处c-n弱的伸缩振动吸收,1624.33cm-1处n-h变形振动以及在900~650cm-1面外弯曲振动,可归属于-hcn亚胺盐结构;2924.63cm-1处的吸收峰为亚甲基-ch2的伸缩振动。
由环合产物小檗红碱的1h-nmr核磁谱图5可知,δppm:8.19、7.53、7.40、7.25,7.08和6.11(1h,苯环),5.90(2h,-o-c-o-),3.35,3.15和3.09(2h,-ch2-),3.84(3h,-och3),2.50(五重峰,氘代dmso溶剂峰),4.4,4.6(h,-oh)。
- 还没有人留言评论。精彩留言会获得点赞!