本发明属于抗癌药物设计、合成领域,具体涉及一种水溶性阳离子型光敏剂及其制备和应用。
背景技术:
肿瘤是目前世界上死亡率最高的疾病之一。光动力治疗(photodynamictherapy,简称pdt)是一种正处在发展中的治疗肿瘤与非肿瘤疾病的新技术,与化疗、放疗等手段相比,光动力治疗具有无耐药性、选择性高、对正常组织损伤小等优点,并且可以和其他成熟的治疗方法相结合使用。光敏剂是影响光动力治疗效果的关键因素,其可以选择性富集在肿瘤组织中,降低对周围正常组织的损伤,产生理想的生物学效应。目前临床上主要使用血卟啉、叶绿素类等传统光敏剂,这类光敏剂组成成分不确定,最大吸收波长在短波长附近,组织穿透能力较弱,光动力治疗时容易造成皮肤光毒性,不是理想的光敏剂。
氟硼二吡咯类衍生物因具有良好的光热稳定性、高的摩尔消光系数、高的单线态氧量子产率、结构易于修饰等优异性能,是具有潜在应用价值的第二代抗癌光敏剂。但是大多数已经报道的氟硼二吡咯光敏剂只能溶于有机溶剂,而生物体系的研究主要是在水溶液的环境下进行,所以氟硼二吡咯光敏剂的疏水性极大地限制了其在实际中的应用。研究表明,光敏剂的亲水性与否将会直接影响光敏剂在生物体内的给药方式及其在细胞内的生理分布。许多光敏剂由于其水溶性太差,导致药物进入到生物体内后被蛋白所吸附而无法经血液输送至肿瘤组织,因而无法起到杀死肿瘤的作用。大量研究表明,带有正电荷取代基的光敏剂往往倾向于在细胞的线粒体中聚集,而线粒体是细胞的能量中枢,经光照后很快引起细胞的凋亡。基于此,本发明尝试制备季铵盐取代的氟硼二吡咯阳离子型光敏剂,期望得到具有良好水溶性的光敏剂,并研究其对肿瘤细胞的光动力杀伤效果。
技术实现要素:
本发明的目的在于提供一种水溶性阳离子型光敏剂及其制备和应用。首先以4-(3,6,9-三氧杂-1-癸氧基)苯甲醛、2,4-二甲基吡咯等为原料制备三乙二醇单甲醚取代的氟硼二吡咯母核;然后在其2,6位引入两个重原子碘,增加其三线态和单线态氧量子产率;再将所得化合物进一步与哌嗪取代的醛进行缩合反应,将其共轭体系扩大,使其吸收移动至红光区;最后将其与碘甲烷作用,获得水溶性阳离子型氟硼二吡咯光敏剂。分别以人肝癌细胞hepg2和人宫颈癌细胞hela为受试细胞株,展开了其体外抗癌活性研究,筛选出适合于分子光动力治疗的前药。本发明合成的化合物结构单一,不存在异构体,产品容易提纯;合成方法比较简单,副反应少,产率较高,原料易得,成本低,有利于工业化生产。
为实现上述目的,本发明采用如下技术方案:
一种水溶性阳离子型光敏剂,所述的光敏剂为氟硼二吡咯季铵盐,化学结构式为:,r为。
所述水溶性抗癌光敏剂的制备方法是以4-(3,6,9-三氧杂-1-癸氧基)苯甲醛()(该化合物的合成参考文献:journaloforganicchemistry,2005,70,7065-7079)、2,4-二甲基吡咯、2,3-二氯-5,6-二氰对苯醌(ddq)、三乙胺(et3n)和三氟化硼乙醚(bf3·et2o)为起始原料,合成化合物x();
然后以x、i2和hio3为起始原料,合成碘代氟硼二吡咯衍生物y();再以y、(该化合物的合成参考文献:journalofmedicinalchemistry,2014,57,5579-5601)、哌啶和冰乙酸为起始原料,合成化合物z();最后以化合物z和碘甲烷为起始原料,合成所述季铵盐取代的氟硼二吡咯衍生物ⅰ。其具体步骤为:
(1)将4-(3,6,9-三氧杂-1-癸氧基)苯甲醛()、2,4-二甲基吡咯按摩尔比为1:2-4加入无水二氯甲烷中,再加入1~3滴三氟乙酸,所得溶液于室温下反应12~24小时,然后加入以4-(3,6,9-三氧杂-1-癸氧基)苯甲醛量计的1当量的2,3-二氯-5,6-二氰对苯醌,继续反应4小时后,三乙胺和三氟化硼乙醚按当量(以4-(3,6,9-三氧杂-1-癸氧基)苯甲醛量计)为5~10:10~20在冰浴条件下加入反应体系中,室温搅拌过夜后结束反应,用硅藻土过滤除去黑色固体,依次用饱和碳酸氢钠和水洗涤粗产品,随后以石油醚和二氯甲烷为洗脱剂,硅胶柱层析分离得到化合物x;
(2)将化合物x、i2、hio3按摩尔比为1:2.5:2加入乙醇中,氮气保护下60℃反应1~3小时;反应结束后减压蒸去溶剂,随后以石油醚和二氯甲烷为洗脱剂,硅胶柱层析分离得到碘代氟硼二吡咯衍生物y;
(3)将化合物y、按摩尔比1:4-10加入到干燥的甲苯中,再将哌啶与冰乙酸按当量(以化合物y的量计)30~40:40~50加入到反应混合液中,最后加入催化量的高氯酸镁,装上分水器,回流1-3小时;反应结束后用ch2cl2和水萃取;有机层用无水na2so4干燥后减压旋干;然后以二氯甲烷和甲醇为洗脱剂,经硅胶柱层析分离得到化合物z;
(4)将化合物z和碘甲烷按摩尔比1:10-40溶解于无水n’n-二甲基甲酰胺(dmf)中,氮气保护下室温搅拌反应过夜;反应结束后,往反应液中加入大量的二氯甲烷,静置60分钟,固体析出并粘附在瓶壁上,过滤得到产物水溶性阳离子型光敏剂i。
所述的水溶性阳离子型氟硼二吡咯衍生物,用于制备抗癌光敏剂,用于光动力治疗。
光动力治疗(photodynamictherapy,pdt)是一种新型的肿瘤治疗方法。其基本要素包括光敏剂、可见光和氧。光敏剂作为能量的载体与反应的桥梁,在光动力治疗中起着决定性的作用。理想的光敏剂最好满足以下几条:组分单一,结构明确,性质稳定;特异靶向性强,能在到达靶组织后迅速达到最高浓度;体内清除时间短,在光照时具有较强的光毒性,而在黑暗情况下无毒副作用;光敏化能力强,单线态氧量子产率高;最长激发波长位于近红外区,在光动力治疗窗口(650-800nm)有较强吸收。氟硼二吡咯衍生物因具有优良的光物理、光化学性质(较高的摩尔消光系数和荧光量子产率,对化学环境较不敏感,无漂白活性等)而成为理想的光敏剂之一。本发明合成了一个在近红外区具有较强吸收的氟硼二吡咯衍生物,母体通过用碘修饰从而增加了其单线态氧量子产率和光毒性,此外在氟硼二吡咯环上引入两个个哌嗪基团并将其中的三级胺进一步甲基化,获得氟硼二吡咯季铵盐,这有利于光敏剂在生物介质中的溶解性,提高药物的生物利用度。
本发明的显著优点在于:
(1)该氟硼二吡咯衍生物含四个四级铵离子,具有较好的水溶性,这有利于制备药物制剂和提高药物的生物利用度;
(2)该氟硼二吡咯衍生物为阳离子型光敏剂,能富集于癌细胞中的线粒体中,有利于光照条件下细胞的凋亡;
(3)该氟硼二吡咯衍生物经化学修饰,其最大吸收和发射位于红光区,组织穿透能力较强,光动力治疗时不易造成皮肤光毒性,是较为理想的光敏剂;
(4)相比卟啉光敏剂,其光稳定性更高;
(5)目标化合物结构单一,不存在异构体,产物容易纯化;
(6)合成方法简单,只需要几个步骤就可以完成,副反应少,原料易得,成本低,有利于工业化生产。
附图说明
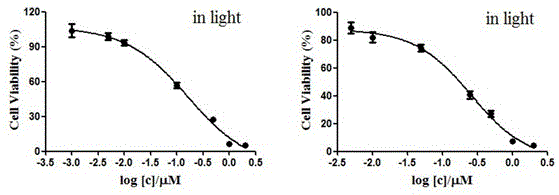
图1为光照条件下,季铵盐取代的氟硼二吡咯衍生物对hepg2(左)和hela(右)细胞的杀伤曲线。
具体实施方式
具有光动力抗癌活性的水溶性阳离子型氟硼二吡咯衍生物的具体制备过程包括:
(1)将4-(3,6,9-三氧杂-1-癸氧基)苯甲醛()、2,4-二甲基吡咯按摩尔比为1:2-4加入无水二氯甲烷中,再加入1~3滴三氟乙酸,所得溶液于室温下反应12~24小时,然后加入以化合物4-(3,6,9-三氧杂-1-癸氧基)苯甲醛量计的1当量的2,3-二氯-5,6-二氰对苯醌,继续反应4小时后,三乙胺和三氟化硼乙醚按当量为5~10:10~20在冰浴条件下加入反应体系中,室温搅拌过夜后结束反应,用硅藻土过滤除去黑色固体,依次用饱和碳酸氢钠和水洗涤粗产品,随后以石油醚和二氯甲烷为洗脱剂,硅胶柱层析分离得到橙红色固体化合物x,其结构式为:,产率18-25%;
(2)将化合物x、i2、hio3按摩尔比为1:2.5:2加入乙醇中,氮气保护下60℃反应1~3小时;反应结束后减压蒸去溶剂,随后以石油醚和二氯甲烷为洗脱剂,硅胶柱层析分离得到红色固体化合物y,其结构式为:,产率60-72%;
(3)将化合物y、按摩尔比1:4-10加入到干燥的甲苯中,再将哌啶与冰乙酸按当量(以化合物y的量计)30~40:40~50加入到反应混合液中,最后加入催化量的高氯酸镁,装上分水器,回流1-3小时;反应结束后用ch2cl2和水萃取;有机层用无水na2so4干燥后减压旋干;然后以二氯甲烷和甲醇为洗脱剂,经硅胶柱层析分离得到绿色固体化合物z,其结构式为:,产率26-34%;
(4)将化合物z和碘甲烷按摩尔比1:10-40溶解于无水n’n-二甲基甲酰胺(dmf)中,氮气保护下室温搅拌反应过夜;反应结束后,往反应液中加入大量的二氯甲烷,静置60分钟,固体析出并粘附在瓶壁上,过滤得到季铵盐取代的氟硼二吡咯,其结构式为:(r为),产率79-92%。
以下实施例进一步阐述本发明,但是本发明不仅限于此。
实施例1
(1)将4-(3,6,9-三氧杂-1-癸氧基)苯甲醛()(3.50g,13.0mmol)和2,4-二甲基吡咯(2.97g,31.3mmol)用无水二氯甲烷(100ml)充分溶解,加2滴三氟乙酸(tfa),室温搅拌反应12h。准确称取一定量的2,3-二氯-5,6-二氰对苯醌(ddq)(2.96g,13.0mmol)用无水二氯甲烷(200ml)超声振荡至完全溶解后加入反应液中,所得混合液继续室温搅拌反应4h,然后在冰浴条件下依次滴加三乙胺(18ml,129.5mmol)和三氟化硼乙醚(18ml,145.8mmol),待三氟化硼乙醚滴加完后,氮气保护下室温反应过夜。反应结束后,先用硅藻土过滤,滤液依次用饱和碳酸氢钠溶液和水洗涤三次,合并有机层,干燥后减压旋干。以二氯甲烷-石油醚(2:1,v/v)为淋洗剂,硅胶柱色谱纯化后得到橙红色固体产物()1.40g,23%。1hnmr(400mhz,cdcl3):δ=7.16(d,j=8.4hz,2h,arh),7.02(d,j=8.4hz,2h,arh),5.97(s,2h,pyrrole-h),4.18(t,j=4.4hz,2h,och2),3.91(t,j=4.4hz,2h,och2),3.76-3.79(m,2h,och2),3.70-3.73(m,2h,och2),3.66-3.69(m,2h,och2),3.56-3.58(m,2h,och2),3.39(s,3h,och3),2.55(s,6h,ch3),1.42(s,6h,ch3)。hrms(esi):c26h33bf2n2o4理论计算值(m/z[m+h]+)为487.2580,实际所测值为487.2586。
(2)将步骤(1)所得化合物(0.20g,0.42mmol)加入到150ml无水乙醇中;充分搅拌至其完全溶解后,准确称取一定量的碘(0.27g,1.04mmol)加入至反应液中,充分搅拌使其完全溶解;再准确称取碘酸(0.15g,0.83mmol)用少量的水溶解后加入至反应液中,氮气保护下60℃反应2h;反应结束后,除去溶剂,剩余残留物用二氯甲烷-石油醚(2:1,v/v)为洗脱剂,硅胶色谱柱纯化后得到红色固体产物()0.20g,68%。1hnmr(400mhz,cdcl3):δ=7.12(d,j=8.8hz,2h,arh),7.04(d,j=8.4hz,2h,arh),4.20(t,j=4.8hz,2h,och2),3.88(t,j=4.8hz,2h,och2),3.72-3.74(m,2h,och2),3.68-3.70(m,2h,och2),3.62-3.64(m,2h,och2),3.56-3.58(m,2h,och2),3.39(s,3h,och3),2.64(s,6h,ch3),1.43(s,6h,ch3)。hrms(esi):c26h31bf2i2n2o4理论计算值(m/z[m+h]+)为739.0513,实际所测值为739.0522。
(3)将步骤(2)所得化合物(0.15g,0.21mmol)、(0.35g,1.40mmol)用无水甲苯(40ml)超声震荡至充分溶解,用移液管依次移取哌啶(0.6ml,6.55mmol)、冰醋酸(0.5ml,8.73mmol)至上述溶液中,再向反应液中加入少量的无水高氯酸镁,避光条件下加热至回流反应2h,反应过程中产生的水用分水器除去。反应结束后,往反应液中加入100ml二氯甲烷,用水洗涤三次,收集有机层,干燥后减压旋干。以二氯甲烷-甲醇(30:1,v/v)为洗脱剂,硅胶色谱柱纯化后得到绿色固体产物()0.073g,31%。1hnmr(400mhz,cdcl3):δ=8.11(d,j=16.8hz,2h,ch=ch),7.58(d,j=8.0hz,4h,arh),7.56(d,j=16.8hz,2h,ch=ch),7.14(d,j=8.4hz,2h,arh),7.05(d,j=8.4hz,2h,arh),6.93(d,j=8.0hz,4h,arh),4.20(t,j=4.8hz,2h,och2),4.15(t,j=5.6hz,4h,och2),3.93(t,j=4.8hz,2h,och2),3.78(t,j=4.8hz,2h,och2),3.72(t,j=5.2hz,2h,och2),3.68(t,j=4.8hz,2h,och2),3.58(t,j=4.4hz,2h,och2),3.40(s,3h,och3),2.84-2.89(br,20h,nch2),2.58(s,6h,nch3),1.49(s,6h,ch3)。hrms(esi):c54h67bf2i2n6o6理论计算值(m/z[m+2h]2+)为600.1715,实际所测值为600.1714。
(4)将步骤(3)所得化合物(0.050g,0.042mmol)溶解于6ml的无水dmf中,待其完全溶解后,再加入碘甲烷(0.18g,1.30mmol),氮气保护下室温搅拌反应过夜。反应结束后,往反应液中加入大量的二氯甲烷(200ml),静置60min后,固体析出并粘附在瓶壁上,过滤,真空干燥(60℃)得到绿色固体产物(,r为)0.059g,88%。1hnmr(400mhz,dmso-d6):δ=8.06(d,j=16.4hz,2h,ch=ch),7.65(d,j=8.4hz,4h,arh),7.43(d,j=16.4hz,2h,ch=ch),7.33(d,j=8.0hz,2h,arh),7.17(d,j=8.4hz,4h,arh),7.15(d,j=8.0hz,2h,arh),4.62(br,4h,och2),4.19(br,8h,och2),3.96(br,20h,nch2),3.80(t,j=4.8hz,4h,och2),3.61(s,3h,och3),3.54(s,6h,nch3),3.24(s,12h,nch3),1.49(s,6h,ch3)。hrms(esi):[c58h79bf2i2n6o6]4+理论计算值(m/z[m]4+)为314.6048,实际所测值为314.6042。
应用实例1
对水溶性阳离子型氟硼二吡咯衍生物(,r为)的离体光动力抗癌活性进行了研究,该实验能为以后在体实验提供一定的参考价值,具有较重要的意义。光敏剂的细胞毒性实验通常包括光毒性和暗毒性两部分,采用mtt法(四氮唑盐还原法)测定。检测原理是活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐)还原为不溶于水的蓝紫色结晶甲瓒并沉积在细胞中,而死细胞中并无琥珀酸脱氢酶,因此不会产生甲瓒。用dmso(二甲基亚砜)溶解活细胞产生的甲瓒,用酶标仪测定其在570nm波长处的吸收值,可间接反映活细胞数量。在一定细胞数范围内,甲瓒形成的量与活细胞数成正比。
mtt实验方法和操作:取生长状态良好的人肝癌细胞hepg2和人宫颈癌细胞hela,用0.25%胰酶消化传代,用rpmi-1640培养基(含10%小牛血清)配制7×104cells/ml细胞悬浮液,将该细胞悬浮液按每孔100μl(约含7000个癌细胞)接种于96孔培养板,置37℃、5%co2培养箱内培养过夜,细胞贴壁后加药;实验设空白对照组(空白对照组是指对照组除了不加药物溶液外,其他条件与受试样品组一致)和溶剂对照组(溶剂对照组是指对照组不加细胞,其他条件与受试样品组一致)。将水溶性阳离子型氟硼二吡咯衍生物预先配制为dmso(含5%蓖麻油)储备液,所有药液配制后均经有机膜过滤(0.22µm),使用时药物溶液用水稀释为不同浓度。每个浓度设定6个平行孔,每孔加入100µl不同浓度的药物后置于培养箱内孵育。光毒实验:24小时后,去除含药液的培养基,用pbs洗涤三次后换上100µl新鲜培养基,然后用红色led灯(波长为660nm)对细胞进行照射,照射能量密度为1.5j·cm-2。光照完毕,将96孔板重置于37℃、5%co2的细胞培养箱内,继续培养。暗毒实验则在换完新鲜培养基后直接放入培养箱中继续培养,操作过程应避免光照,24h后,除空白对照组外,每孔均加入10μl预先配置好的mtt溶液(4mg·ml-1),37℃孵育4小时,4小时后小心弃去上清液,每孔加入100μldmso溶解甲瓒颗粒,轻度震荡使甲瓒完全溶解后,用酶标仪测定570nm波长下od值。
我们采用mtt法测定了实施例1制备的季铵盐取代的氟硼二吡咯衍生物,在光照和无光照条件下,对人肝癌细胞hepg2以及人宫颈癌细胞hela的杀伤效果,光照波长为660nm,光照能量密度为1.5j·cm-2。数据由三次独立的平行实验得到,以mean±sem方式处理。由实验数据可知:在无光照条件下,季铵盐取代的氟硼二吡咯衍生物对两种细胞均没有任何杀伤作用;而在光照条件下,其对人肝癌细胞hepg2和人宫颈癌细胞hela表现出很强的体外毒性,其半数抑制浓度(ic50值)分别为0.16μm和0.26μm(见图1和表1)。
表1季铵盐取代的氟硼二吡咯衍生物对hepg2和hela细胞的ic50值
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。