噻吩并吡啶杂环化合物及其衍生物的制备方法与流程
本发明涉及一种噻吩并吡啶杂环化合物及其衍生物的制备方法。
背景技术:
4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐用作抗血栓药氯吡格雷的中间体,价格低廉,易于获得。而噻吩并[3,2-c]吡啶价格昂贵,在国内售价为1克约1500元;而通过4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐为原料制备的2-溴噻吩并[3,2-c]吡啶的售价为5克9800元;2,3-二溴[3,2-c]吡啶化合物在国内没有供应商(通过chemicalbook查询供应商,价格为国内供应商售价)。制备方法中所采用的原料4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐价廉易得;各个步骤的反应条件温和,后处理操作简单,反应收率较高。可见,本发明的制备方法操作便捷,产物成本低,符合工业化生产的要求,且噻吩并吡啶杂环化合物及其衍生物在离子液体、离子液晶、有机半导体材料中有广泛的应用。因此本发明旨在开发出一种简单便捷、适合工业化生产的噻吩并吡啶杂环化合物及其衍生物的制备方法。
技术实现要素:
本发明提供了一种噻吩并吡啶杂环化合物及其衍生物的制备方法:
一、一种噻吩并[3,2-c]吡啶杂环化合物i的制备方法,包括步骤如下:(1)以4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐在碱性条件下发生中和反应得到化合物2,(2)化合物2与二氧化锰在溶剂中搅拌回流发生芳香化反应得到化合物i,见以下反应路线:
二、一种2-溴噻吩并[3,2-c]吡啶化合物的制备方法,包括步骤如下:(1)以4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐与溴单质在溶剂中发生溴代反应得到化合物3,(2)化合物3在碱性条件下发生中和反应得到化合物4,(3)化合物4与二氧化锰在溶剂中搅拌回流发生芳香化反应得到化合物ii,见以下反应路线:
三、一种2,3-二溴[3,2-c]吡啶化合物的制备方法,包括步骤如下:(1)以4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐与溴单质在溶剂中发生溴代反应得到化合物5,(2)化合物5在碱性条件下发生中和反应得到化合物6,(3)化合物4与二氧化锰在溶剂中搅拌回流发生芳香化反应得到化合物iii,见以下反应路线:
本发明的优点在于:(1)制备方法中所采用的原料4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐以及反应所需试剂均为常规试剂,价廉易得;(2)各个步骤的反应条件温和,后处理操作简单,(3)反应收率较高,且产物纯度高。可见,本发明的制备方法操作便捷,产物成本低,符合工业化生产的要求。
附图说明
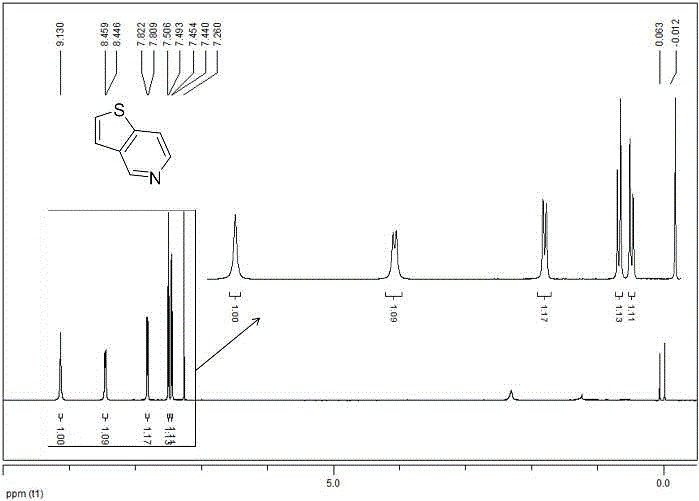
图1为实施例1中所获化合物i的核磁共振氢谱。
图2为实施例2中所获化合物ii的核磁共振氢谱。
图3为实施例3中所获化合物iii的核磁共振氢谱。
具体实施方式
以下将通过具体实施例进行进一步阐述本发明,但并不用于限制本发明的保护范围。在不脱离本发明构思的前提下,本领域技术人员可在权利要求范围内对制备方法和使用仪器作出改进,这些改进也应视为本发明的保护范围。
实施例1、化合物i的制备
将4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐(1.0g,5.69mmol)和氢氧化钠(228mg,5.69mmol)加入到水溶液(20ml)中,搅拌反应2小时后,通过二氯甲烷萃取,减压蒸馏除溶剂后;加入事先活化好的二氧化锰(3.5g,39.8mmol),并加入甲苯(5ml)做溶剂,在加热(130oc)搅拌回流的条件下反应12-24小时,以薄层色谱法确定原料反应完全则终止反应;反应结束后,用漏斗过滤二氧化锰,收集有机相,减压蒸馏除去溶剂后得到残留物,通过硅胶柱色谱法(溶剂:二氯甲烷)纯化,得到化合物i(650mg),收率84%,为白色固体;以下的数据表明,采用以上本例方法制得的产物确是化合物i(噻吩并[3,2-c]吡啶)。
核磁氢谱1hnmr(cdcl3,tms,400mhz)δ:9.13(s,1h,arh),8.45(d,j=5.2hz,1h,arh),7.82(d,j=5.2hz,1h,arh),7.50(d,j=5.2hz,1h,arh),7.45(d,j=5.6hz,1h,arh)。
实施例2、化合物ii的制备
将4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐(1.0g,5.69mmol)加入配有搅拌子的圆底烧瓶中,加入三氯甲烷(50ml),再用恒压滴液漏斗将溶于三氯甲烷(15ml)的溴单质(1.0g,6.29mmol)缓慢滴加到圆底烧瓶中,滴加完后,搅拌反应过夜,反应结束后,过滤生成的固体;将固体和氢氧化钠(228mg,5.69mmol)加入到二氯甲烷(20ml)和水(10ml)的混合溶剂中,搅拌反应2小时后,分液,通过二氯甲烷萃取,减压蒸馏除溶剂后;加入事先活化好的二氧化锰(2.5g,27.5mmol),并加入甲苯(5ml)做溶剂,在加热(130oc)搅拌回流的条件下反应12-24小时,以薄层色谱法确定原料反应完全则终止反应;反应结束后,用漏斗过滤二氧化锰,收集有机相,减压蒸馏除去溶剂后得到残留物,通过硅胶柱色谱法(溶剂:二氯甲烷:乙酸乙酯=10:1)纯化,得到化合物ii(804mg),收率66%;以下的数据表明,采用以上本例方法制得的产物确是化合物ii(2-溴噻吩并[3,2-c]吡啶)。
核磁氢谱1hnmr(cdcl3,tms,400mhz)δ:8.98(s,1h,arh),8.43(d,j=5.6hz,1h,arh),7.67(d,j=5.6hz,1h,arh),7.43(s,1h,arh)。
实施例3、化合物iii的制备
将4,5,6,7-四氢噻吩[3,2-c]吡啶盐酸盐(1.0g,5.69mmol)加入配有搅拌子的圆底烧瓶中,加入三氯甲烷(50ml),再用恒压滴液漏斗将溶于三氯甲烷(15ml)的溴单质(1.8g,11.4mmol)滴加到圆底烧瓶中,滴加完后,搅拌加热(80oc)反应过夜,反应结束后,过滤生成的固体,将固体和氢氧化钠(228mg,5.69mmol)加入到二氯甲烷(20ml)和水(10ml)的混合溶剂中,搅拌反应2小时后,分液,通过二氯甲烷萃取,减压蒸馏除溶剂后得到残留物;通过硅胶柱色谱法(溶剂:二氯甲烷:乙醇=10:1)纯化,得到化合物6,然后加入事先活化好的二氧化锰(2.5g,27.5mmol),并加入甲苯(5ml)做溶剂,在加热(130oc)搅拌回流的条件下反应12-24小时,以薄层色谱法确定原料反应完全则终止反应;反应结束后,用漏斗过滤二氧化锰,收集有机相,减压蒸馏除去溶剂后得到残留物,通过硅胶柱色谱法(溶剂:二氯甲烷:乙酸乙酯=10:1)纯化,得到化合物ii(965mg),收率57%;以下的数据表明,采用以上本例方法制得的产物确是化合物iii(2,3-二溴[3,2-c]吡啶)。
核磁氢谱1hnmr(cdcl3,tms,400mhz)δ:9.01(s,1h,arh),8.54(d,j=4.8hz,1h,arh),7.66(d,j=5.2hz,1h,arh)。
- 还没有人留言评论。精彩留言会获得点赞!