一种制备、纯化HOPS复合物蛋白的方法与流程
本发明涉及一种制备、纯化hops复合物蛋白的方法。
背景技术:
蛋白质的纯化制备是一项重要的操作技术,在生物化学研究中应用广泛。膜蛋白是构成生物膜系统的关键组分,同时在多种细胞生命活动中发挥重要作用,但由于膜蛋白具有疏水性强、丰度低等理化性质,其提取制备一直存在着效率低、纯度低等问题。
同型融合和蛋白质分选(homotypicfusionandvacuoleproteinsorting,hops)复合物由六种79-123kda的蛋白组成,包括vps11、vps16、vps18、vps33、vps39和vps41。hops复合物在进化上很保守,参与细胞内的膜泡运输,对于酵母中吞噬泡与液泡,以及哺乳动物细胞中自噬体与溶酶体融合的过程至关重要。
细胞自噬广泛存在于真核生物中,是细胞内物质代谢的重要途径,在生长发育和多种疾病中发挥着关键作用。自噬体包裹着错误折叠蛋白、损伤的细胞器等待降解物质,通过与溶酶体融合,形成自噬溶酶体,完成底物的降解,因此膜融合是自噬过程的关键步骤。hops复合物、snare复合物、rabgtp酶等介导了自噬体-溶酶体融合的过程。其中,hops复合物通过与snare蛋白stx17相互作用促进自噬体与溶酶体的膜融合过程。因此,纯化、制备hops蛋白复合物,将有助于揭示自噬及其他膜融合过程的机制。
hops是一种膜结合蛋白复合物,现有技术中,膜蛋白的纯化制备是一大难点:膜蛋白的疏水性使其容易沉降和聚集,且对于溶解条件要求苛刻;和胞质蛋白相比,膜蛋白的丰度低;复杂的翻译后修饰、跨膜区较少的酶切位点等特点都增加了其提取、纯化的难度。这就导致了膜蛋白的纯化需要大量的起始细胞,且纯化过程繁琐,获得的蛋白纯度不够等问题。
技术实现要素:
针对这些缺点,本发明提供了一种较为简便、高效地在体外诱导表达并纯化hops蛋白的方法,可以得到高纯度的膜蛋白。
为了达到上述目的,本发明所采用的技术方案是:一种制备、纯化hops复合物蛋白的方法,其特征在于,包括如下步骤:
1)设计用于扩增组成人hops复合体的6种基因的编码区的引物;所述的6种基因为:人vps11基因、人vps18基因、人vps39基因、人vps16基因、人vps33基因、人vps41基因;分别扩增得到人vps11基因、人vps18基因、人vps39基因、人vps16基因、人vps33基因、人vps41基因片段;
2)分别构建pet-his-nusa-tev-ha-vps11、pet-his-nusa-tev-ha-vps18、pet-his-nusa-tev-ha-vps39的重组质粒;分别构建pgex4t-1-gst-tev-flag-vps16、pgex4t-1-gst-tev-flag-vps33、pgex4t-1-gst-tev-flag-vps41的重组质粒;
3)将步骤2)构建的6种重组质粒分别转化大肠杆菌细胞,再进行融合蛋白的诱导表达和分离纯化;
其中,针对pet-his-nusa-tev-ha-vps11、pet-his-nusa-tev-ha-vps18、pet-his-nusa-tev-ha-vps39,iptg诱导表达,且在22°c条件下诱导16h,iptg诱导终浓度为0.2mmol/l,然后采用ni柱亲和层析的方法纯化蛋白;
针对pgex4t-1-gst-tev-flag-vps16、pgex4t-1-gst-tev-flag-vps33、pgex4t-1-gst-tev-flag-vps41,iptg诱导表达,iptg诱导终浓度为0.2mmol/l,诱导温度为28°c,诱导时间为5h,然后采用谷胱甘肽琼脂糖树脂亲和层析的方法纯化蛋白。
用于扩增组成人hops复合体的6种基因的编码区的引物包括:
a.用于扩增人vps11基因的上下游引物
vps11基因上游引物:5'-ctggtgagaacctgtacttccaatccggatcctacccctacgacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagtttctgctcaagctt
ttaagtgcccctcctggagtgc-3';
b.用于扩增人vps18基因的上下游引物
vps18基因上游引物:5'-ctggtgagaacctgtacttccaatccggatcctacccctacgacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagtttctgctcaagct
tctacagccaactgagctgctcctc-3';
c.用于扩增人vps39基因的上下游引物
vps39基因上游引物:5'-ctggtgagaacctgtacttccaatccggatcctacccctac
gacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagtttctgctcaagcttc
taagtgtcagctgggtttacctc-3';
d.用于扩增人vps16基因的上下游引物
vps16基因上游引物:5'-ccggaattcatggactgctacacggcgaac-3',下游引物:atttgcggccgcctacttcttctgggcttgtgccct-3';
e.用于扩增人vps33基因的上下游引物
vps33基因上游引物:5'-acaagggatccccggaattcatggcggctcatctgtcc-3',下游引物:5'-cagtcagtcacgatgcggccgcctagaaaggtttttccatcag-3';
f.用于扩增人vps41基因的上下游引物
vps41基因上游引物:5'-cgcggatccatggcggaagcagaggagcaggaaac-3',下游引物:5'-atttgcggccgcctattttttcatctccaaaattgcac-3'。
以上序列依次为seqidno.1-12。
针对pet-his-nusa-tev-ha-vps11、pet-his-nusa-tev-ha-vps18、pet-his-nusa-tev-ha-vps39,其融合蛋白的诱导表达和分离纯化具体过程为:37°c,200rpm将细胞培养至600nm波长下的od值在0.9~1.2之间,加入iptg,22°c诱导16h,然后离心弃上清得到菌体,菌体用e.colilysisbuffer重悬,静置30min后冰上进行超声破碎,离心收集上清;提前将ni-nta琼脂糖珠装入ni纯化柱中,用2倍柱体积的平衡液清洗,将菌体上清加入到纯化柱中,4°cip架上旋转摇2h,离心弃流出液,加入2倍柱体积的清洗液清洗,离心弃流出液,加入一定量洗脱液进行洗脱,收集流出液,使用tev酶将his-nusa标签切除,二次过柱,标签蛋白与ni柱结合,而目的蛋白则流出。所述的目的蛋白为ha-vps11、ha-vps18、ha-vps39。
针对pgex4t-1-gst-tev-flag-vps16、pgex4t-1-gst-tev-flag-vps33、pgex4t-1-gst-tev-flag-vps41,其融合蛋白的诱导表达和分离纯化具体过程为:37°c,200rpm至600nm波长下的od值在0.4~0.6间,加入iptg,28°c诱导5h,然后离心弃上清得到菌体;菌体用e.colilysisbuffer重悬,静置30min后冰上进行超声破碎,离心收集上清,向上清中加入一定量的glutathionesepharose4b,4°c旋转摇过夜,次日取出离心弃上清,将beads清洗3次,离心去除清洗液,最后加入适量e.colilysisbuffer和tev酶,4°c旋转结合2h后,离心收集上清即得最终纯化好的融合蛋白。所述的融合蛋白为flag-vps16、flag-vps33、flag-vps41。
相对于现有技术,本发明有如下有益效果:
(1)本发明实现了膜结合蛋白hops复合物六种不同组分的分离纯化,说明该方法在纯化不同的膜蛋白时有通用性和稳定性的特点。
(2)本发明利用原核表达系统进行蛋白纯化,易于操作,经济、可行。
(3)本发明利用亲和层析的方法完成蛋白的分离,通过westernblot的验证,该方法得到的蛋白有很好的特异性。
综合以上三点,本发明可以简便地获得高特异性的hops复合物蛋白,为膜蛋白的分离纯化提供了更好的方法,能够为生物化学等基础研究提供支持。
附图说明
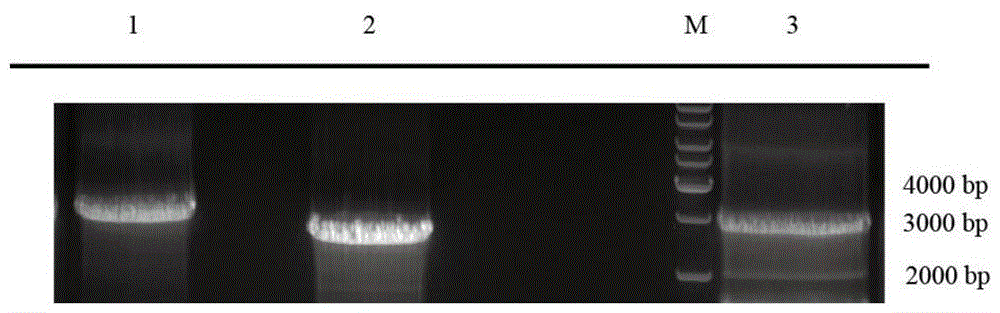
图1.vps18、vps39和vps11基因的pcr扩增,m:dnamarker;1:vps18基因的pcr产物;2:vps39基因的pcr产物;3:vps11基因的pcr产物;
图2.vps16、vps41和vps33基因的pcr扩增,m:dnamarker;1:vps16基因的pcr产物;2:vps41基因的pcr产物;3:vps33基因的pcr产物;
图3.pet-his-nusa-tev-ha-vps11、vps39、vps18重组质粒菌落pcr鉴定结果,m:dnamarker;1-3:pet-his-nusa-tev-ha-vps11重组质粒菌落pcr鉴定;4-6:pet-his-nusa-tev-ha-vps39重组质粒菌落pcr鉴定;7-9:pet-his-nusa-tev-ha-vps18重组质粒菌落pcr鉴定;
图4.pgex4t-1-gst-tev-flag-vps16、vps41、vps33重组质粒菌落pcr鉴定结果,m:dnamarker;1-2:pgex4t-1-gst-tev-flag-vps16重组质粒菌落pcr鉴定;3-5:pgex4t-1-gst-tev-flag-vps41重组质粒菌落pcr鉴定;6-8:pgex4t-1-gst-tev-flag-vps33重组质粒菌落pcr鉴定;
图5.ha-vps11、ha-vps18、ha-vps39融合蛋白的sds-page与westernblot法分析,m:marker;1:ha-vps11蛋白;2:ha-vps18蛋白;3:ha-vps39蛋白;
图6.flag-vps16、flag-vps33、flag-vps41融合蛋白的sds-page与westernblot法分析,m:marker;1:flag-vps16蛋白;2:flag-vps33蛋白;3:flag-vps41蛋白。
具体实施方式
下面结合具体实例对本发明做进一步的描述。
(1)本发明中所用试剂、耗材及生物材料的具体来源
pgex4t-1原核表达质粒由本实验室保存;pet-28a(+)改造的pet-his-nusa原核表达质粒由南京农业大学理学院万群实验室惠赠。e.colidh5α、bl21(de3)感受态细胞购自上海唯地生物技术公司;pcr试剂盒、t4连接酶购自takara公司;treliefsosoocloningkit购自南京擎科生物技术有限公司;限制性核酸内切酶ecori、bamhi、hindiii和noti购自neb公司;胶回收试剂盒、dnamarker购自genstar公司;质粒小提试剂盒购自omega公司;tae、氨苄青霉素(ampicillin)、卡那青霉素(kanamycin)、异丙基硫代半乳糖苷(iptg)和考马斯快速染色液均购自索莱宝公司;引物合成及dna序列测序均由南京金斯瑞生物科技有限公司完成;glutathionesepharose4b购自美国ge公司;nifocurose6ff购自武汉汇研生物科技股份有限公司;鼠单克隆抗flag抗体购自sigma公司;鼠单克隆抗ha抗体、hrp标记的山羊抗鼠igg、山羊抗兔igg购自sba公司;化学发光试剂购自bio-rad公司。
(2)扩增组成人hops复合体的6种基因的编码区
1)人vps11基因的上下游引物
根据vps11基因序列合成引物;上游引物:5'-ctggtgagaacctgtacttccaatccg
gatcctacccctacgacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagttt
ctgctcaagcttttaagtgcccctcctggagtgc-3'(下划线部分分别为bamhi和hindiii的酶切位点)。
2)人vps18基因的上下游引物
根据vps18基因序列合成引物;上游引物:5'-ctggtgagaacctgtacttccaatccg
gatcctacccctacgacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagttt
ctgctcaagcttctacagccaactgagctgctcctc-3'(下划线部分分别为bamhi和hindiii的酶切位点)。
3)人vps39基因的上下游引物
根据vps39基因序列合成引物;上游引物:5'-ctggtgagaacctgtacttccaatccg
gatcctacccctacgacgtgcccgacta-3',下游引物:5'-tcctcttcagagatgagttt
ctgctcaagcttctaagtgtcagctgggtttacctc-3'(下划线部分分别为bamhi和hindiii的酶切位点)。
4)人vps16基因的上下游引物
根据vps16基因序列合成引物;上游引物:5'-ccggaattcatggactgctacacggc
gaac-3',下游引物:atttgcggccgcctacttcttctgggcttgtgccct-3'(下划线部分分别为ecori和noti的酶切位点)。
5)人vps33基因的上下游引物
根据vps33基因序列合成引物;上游引物:5'-acaagggatccccggaattcatggcg
gctcatctgtcc-3',下游引物:5'-cagtcagtcacgatgcggccgcctagaaaggttttt
ccatcag-3'(下划线部分分别为ecori和noti的酶切位点)。
6)人vps41基因的上下游引物
根据vps41基因序列合成引物;上游引物:5'-cgcggatccatggcggaagcagagga
gcaggaaac-3',下游引物:5'-atttgcggccgcctattttttcatctccaaaattgcac-3'
(下划线部分分别为bamhi和noti的酶切位点)。
将上述引物序列送南京金斯瑞公司进行合成,以pcdna3.1-ha-vps11、pcdna3.1-ha-vps16、pcdna3.1-ha-vps18、pcdna3.1-ha-vps33、pcdna3.1-ha-vps39、pcdna3.1-ha-vps41为模板,利用primerstar酶扩增目的片段,条件如下:98°c变性10s、58°c退火5s、72°c延伸1min,共30个循环。50μl的pcr产物用1%琼脂糖凝胶电泳检测,并用dna胶回收试剂盒回收目的基因片段。其中vps11全长2826bp,vps18全长2922bp,vps39全长2628bp,vps16全长2520bp,vps33全长1791bp,vps41全长2565bp,经琼脂糖电泳鉴定获得与预期片段一致的6种pcr产物(结果见附图1和附图2)。
(3)构建六种重组质粒并鉴定
用bamhi和hindiii双酶切pet-his-nusa质粒,用ecori和noti、bamhi和noti分别双酶切pgex4t-1-gst质粒,37°c水浴锅酶切过夜。用dna胶回收试剂盒回收酶切后的载体。根据引物设计的不同,用以下两种方法进行连接:
1)无缝连接方式(vps11、vps18、vps33、vps39):用treliefsosoocloningkit(50°c)将线性化载体与pcr产物连接15min后转化至e.colidh5α感受态细胞中,涂板后37°c培养过夜。
2)t4连接方式(vps16、vps41):pcr产物需要双酶切过夜,回收酶切后的pcr产物,与线性化载体连接,16°c连接5h,时间到后转化至e.colidh5α感受态细胞中,涂板后37°c培养过夜。
采用无缝拼接克隆方式构建pgex4t-1-gst-tev-flag-vps33、pet-his-nusa-tev-ha-vps11、pet-his-nusa-tev-ha-vps18、pet-his-nusa-tev-ha-vps39,无缝拼接方式设计的引物特别之处在于载体的末端与引物的末端具有15~20bp的同源碱基,这样所得的pcr产物两端就会带有与线性化载体两端序列相同的碱基;采用t4连接的方式构建pgex4t-1-gst-tev-flag-vps16和pgex4t-1-gst-tev-flag-vps41,设计的上下游引物末端的酶切位点附近都含有保护碱基,这是因为直接暴露在末端的酶切位点不易被识别需要加入保护碱基来提高酶切时酶的活性。此外,由于pgex4t-1-tev-flag-vps33这一重组质粒的目的片段vps33含有ecori、bamhi、noti这三种酶切位点,而线性化pgex4t-1载体恰恰需要这几种酶,t4连接法需要将pcr产物和载体双酶切过夜,这一步骤会直接将vps33这一目的片段酶切成几条片段,因此采用无缝拼接构建该重组质粒。
次日,取出所有平板,挑取单克隆,进行菌落pcr,用琼脂糖凝胶电泳初步鉴定,选择条带位置正确的菌液进行测序鉴定。测序无误后,进行质粒小提。重组质粒菌落pcr鉴定结果见附图3和附图4。将菌落pcr验证正确的菌液送测,正反向测序结果与基因序列基本一致,说明6种组成hops复合体的原核表达重组质粒构建成功。
(4)融合蛋白的诱导表达和分离纯化
将测序正确的6种重组质粒转化至e.colibl21(de3)感受态细胞中,涂板后37°c培养过夜。挑取单克隆于50ml含抗生素的lb培养基中37°c培养过夜,次日扩大培养至500ml液体lb培养基中(含抗生素)。根据所用载体的不同,用以下两种亲和层析的方法进行蛋白纯化和提取:
1)vps18、vps39或vps11诱导表达、ni柱亲和层析:37°c,200rpm将细胞培养至600nm波长下的od值在0.9~1.2间,加入iptg(终浓度为0.2mmol/l),22°c诱导16h,时间到后5000rpm,30min,4°c离心弃上清得到菌体,菌体用e.colilysisbuffer(20mmtris-hclph7.5,500mmnacl,20mmimidazole,1.4mmβ-巯基乙醇,0.05%tween20,pmsf蛋白酶抑制剂)重悬,静置30min后冰上进行超声破碎(每次3min,超声2s停5s,三个循环),12000rpm,30min,4°c离心收集上清。提前将ni-nta琼脂糖珠装入纯化柱中,用2倍柱体积的平衡液(20mmtris-hclph7.5,500mmnacl,10mmimidazole,5%甘油)清洗3~5次,将菌体上清加入到柱子中,4°cip架上旋转摇2h,离心弃流出液,加入2倍柱体积的清洗液(20mmtris-hclph7.5,500mmnacl,20mmimidazole,5%甘油)清洗3~5次,离心弃流出液,加入一定量洗脱液(20mmtris-hclph7.5,500mmnacl,500mmimidazole,5%甘油)进行洗脱,收集流出液,使用tev酶将his-nusa标签切除,二次过柱,标签蛋白与ni柱结合,而目的蛋白则流出。
2)vps16、vps41或vps33诱导表达、谷胱甘肽琼脂糖树脂亲和层析:37°c,200rpm至600nm波长下的od值在0.4~0.6间,加入iptg(终浓度为0.2mmol/l),28°c诱导5h,时间到后5000rpm,30min,4°c离心弃上清得到菌体。菌体用e.colilysisbuffer(20mmtris-hclph7.5,150mmnacl,0.1%β-巯基乙醇,1%tritonx-100,pmsf蛋白酶抑制剂)重悬,静置30min后冰上进行超声破碎(每次3min,超声2s停5s,三个循环),12000rpm,30min,4°c离心收集上清,向上清中加入一定量的glutathionesepharose4b(磁珠),4°c旋转摇过夜,次日取出离心弃上清,将beads清洗3次,离心去除清洗液,最后加入适量e.colilysisbuffer和tev酶,4°c旋转结合2h后,离心收集上清即得最终纯化好的融合蛋白。
用sds-page和westernblot方法检测纯化蛋白,结果见附图5和附图6。
经sds-page和westernblot方法检测得出以下结果:利用ni柱亲和层析方式纯化得到分子量在105kda左右的ha-vps11融合蛋白,108kda左右的ha-vps18融合蛋白及97kda左右的ha-vps39融合蛋白(图5);利用谷胱甘肽亲和层析方式得到分子量在97kda左右的flag-vps16融合蛋白,70kda左右的flag-vps33融合蛋白及98kda左右的flag-vps41融合蛋白(图6)。
综上,在本发明中,我们利用pgex4t-1-gst和pet-his-nusa原核表达载体成功构建出了6种重组质粒,并且采用谷胱甘肽和ni柱两种亲和纯化方法获得vps11、vps16、vps18、vps33、vps39和vps41这6种蛋白组分,通过sds-page的westernblot方法验证了纯化蛋白的正确性。
本发明利用亲和层析的方法,分离纯化了组成hops复合物的六种组分,为膜蛋白的提取提供了一种简便高效的方案。欲保护点如下:针对人hops复合物组分设计的特异性扩增引物序列。蛋白诱导的具体条件,包括iptg的浓度、诱导温度、诱导时间等。
如果仅仅利用pgex4t-1这一载体,采用谷胱甘肽方式纯化这6种蛋白,培养条件是iptg终浓度为0.35mmol/l,诱导温度为37°c,诱导时间为2h,发现蛋白的表达量均不高,并且基本全部形成了包涵体,蛋白不可溶。因为膜蛋白具有暴露的疏水区,表达时易于聚集形成包涵体。因此,改变了培养条件,通过改变iptg诱导终浓度为0.2mmol/l,诱导温度为28°c,诱导时间为5h进行诱导,降低了蛋白的合成效率,提高了蛋白表达量,改善了pgex4t-1-gst-tev-flag-vps16、vps33、vps41这三种融合蛋白的可溶性,从而成功纯化出flag-vps16、flag-vps33、flag-vps41蛋白。但pgex4t-1-gst-tev-flag-vps11、vps18、vps39这三种重组质粒的表达量及可溶性依然没有明显的改变,因此选择了pet-his-nusa载体重新构建质粒pet-his-nusa-tev-ha-vps11、pet-his-nusa-tev-ha-vps18、pet-his-nusa-tev-ha-vps39。nusa是一个分子量约为55kda的蛋白标签,nusa有利于促进重组蛋白的正确折叠,提高蛋白的表达量。由于其自身的可溶性很高,因此作为融合标签与目的蛋白结合后还可以提高目标蛋白的稳定性及可溶性。此载体上含有t7强启动子,为了减慢蛋白质的合成速度,降低形成包涵体的概率,利用iptg诱导表达时采用22°c诱导16h的条件,最终在his-nusa促融标签的作用下利用ni柱亲和层析方式实现vps11、vps18和vps39的可溶性表达,成功纯化出重组蛋白。
需要说明的是,上述实施例不以任何形式限制本发明,凡采用等同替换或等效变换的方式所获得的技术方案,均落在本发明的保护范围内。
序列表
<110>南京农业大学
<120>一种制备、纯化hops复合物蛋白的方法
<130>xhx2019032501
<141>2019-03-25
<160>12
<170>siposequencelisting1.0
<210>1
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>1
ctggtgagaacctgtacttccaatccggatcctacccctacgacgtgcccgacta55
<210>2
<211>54
<212>dna
<213>人工序列(artificialsequence)
<400>2
tcctcttcagagatgagtttctgctcaagcttttaagtgcccctcctggagtgc54
<210>3
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>3
ctggtgagaacctgtacttccaatccggatcctacccctacgacgtgcccgacta55
<210>4
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>4
tcctcttcagagatgagtttctgctcaagcttctacagccaactgagctgctcctc56
<210>5
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>5
ctggtgagaacctgtacttccaatccggatcctacccctacgacgtgcccgacta55
<210>6
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>6
tcctcttcagagatgagtttctgctcaagcttctaagtgtcagctgggtttacctc56
<210>7
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>7
ccggaattcatggactgctacacggcgaac30
<210>8
<211>36
<212>dna
<213>人工序列(artificialsequence)
<400>8
atttgcggccgcctacttcttctgggcttgtgccct36
<210>9
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>9
acaagggatccccggaattcatggcggctcatctgtcc38
<210>10
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>10
cagtcagtcacgatgcggccgcctagaaaggtttttccatcag43
<210>11
<211>35
<212>dna
<213>人工序列(artificialsequence)
<400>11
cgcggatccatggcggaagcagaggagcaggaaac35
<210>12
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>12
atttgcggccgcctattttttcatctccaaaattgcac38
- 还没有人留言评论。精彩留言会获得点赞!