一种铜催化烯基叠氮合成β-三氟甲基-β-羟基-1,2-二苯基丙酮的方法与流程
本发明属于有机合成领域,具体涉及一种铜催化烯基叠氮合成β-三氟甲基-β-羟基-1,2-二苯基丙酮的方法。
背景技术:
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
近年来,有机氟化合物在药物化学中受到了很多关注。众所周知,将氟引入药物可以改变它们的性质,包括物理性质,化学性质和生物活性等。由于氟原子的电负性大,原子半径小,三氟甲基(cf3)具有强吸电性和亲脂性,且其碳-氟键十分稳定,且由于氟与氢原子的大小相近,使含氟药物具有拟态效应,因此,将三氟甲基引入到分子结构中能够显著改变药物的酸性、极性、亲脂性及其化学和生物活性(精细石油化工,2005,(2):46-50;精细化工,2004,21(8):639-640),同时大大的增强了稳定性与脂溶性,代谢能力也不受影响,药物用量少,副作用小(精细化工,2004,21(8):639-640),因此,在温和的反应条件下,将三氟甲基引入有机化合物中是科研人员研究的热点。
β-三氟甲基-β-羟基-1,2-二苯基丙酮是一种实用的化合物,在医药、染料和材料中都有着很重大的应用。由于β-三氟甲基-β-羟基-1,2-二苯基丙酮的重要性,其简单实用合成方法的发展仍引起人们极大兴趣。如2011年,日本保和大学药物化学研究所的higashiyama教授利用et2zn直接催化三氟苯乙酮与酮的羟醛缩合反应制备β-三氟甲基-β-羟基-1,2-二苯基丙酮(synlett,2011,10,1431-1434)。2012年,天津大学化学系的马俊安教授报道了在dhqd生物碱的存在下,β-酮酸与三氟苯乙酮选择性脱羧并发生羟醛缩合反应制备β-三氟甲基-β-羟基-1,2-二苯基丙酮(chem.commun.,2012,48,4308-4310)。2015年,西北师范大学化学化工学院杜正银教授课题组实现了在无溶剂条件下,由苯乙酮和三氟苯乙酮完成了β-三氟甲基-β-羟基-1,2-二苯基丙酮的快速制备。然而目前,β-三氟甲基-β-羟基-1,2-二苯基丙酮的合成还主要是使利用三氟苯乙酮与另一种醛或酮进行醛醇缩合反应,但反应条件繁琐,产物单一,选择性差、产率较低。
技术实现要素:
为了克服上述问题,并同时考虑环保与经济效益,本发明提供了一种简便高效的制备β-三氟甲基-β-羟基-1,2-二苯基丙酮的方法,所述方法以醋酸铜为催化剂,以简便易得的烯基叠氮和三氟苯乙酮为底物,在温和的条件下,利用烯基叠氮β碳官能化,在铜的催化剂作用下通过一锅法实现羟醛反应策略制备β-三氟甲基-β-羟基-1,2-二苯基丙酮类化合物。本发明利用简单廉价的金属铜催化高活性的烯基叠氮β碳官能化,通过羟醛反应,实现了高区域选择性的制备β-三氟甲基-β-羟基-1,2-二苯基丙酮。该方法操作极其简单、底物和催化剂简便易得、底物普适性优良。
为实现上述技术目的,本发明采用的技术方案如下:
一种合成式(i)所示化合物的方法,包括:
将烯基叠氮、三氟苯乙酮混合均匀,在醋酸铜存在下,合成式(i)化合物;
其中,所述式(i)的结构式为:
基团r选自富电子或缺电子的芳基、取代芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基;
基团r1选自富电子或缺电子的芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基。
在一些实施例中,所述基团r为芳基,所述芳基为苯基;
在一些实施例中,所述基团r为取代芳基,所述取代芳基为被选自卤素、c1-c6直链或支链的烷基和c1-c2直链的烷氧基所取代的苯基;所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;所述直链烷基选自c3-c7直链烷基;
在上述实施例中,更为优选地方案为:所述取代芳基为被选自f、cl、br、甲基、乙基、正丙基、叔丁基、正戊基、甲氧基、乙氧基所取代的苯基;
在一些实施例中,所述基团r为芳杂基,所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;
在一些实施例中,所述基团r为直链烷基,所述直链烷基选自辛基,庚基,己基。
在一些实施例中,所述基团r1为芳基,所述芳基为苯基;
在一些实施例中,所述基团r1为取代芳基,所述取代芳基选自卤素、烷基和烷氧基所取代的苯基;
在上述实施例中,更为优选地方案为:所述取代芳基为被选自br、甲基、甲氧基、硝基所取代的苯基;
在一些实施例中,所述基团r1为取代芳基为芳杂基,所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;
在一些实施例中,所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶。
在一些实施例中,所述烯基叠氮的化合物具有式(ii)所示结构:
其中,基团r选自富电子或缺电子的芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基。
在一些实施例中,所述基团r为芳基,所述芳基为苯基;
在一些实施例中,所述r为取代芳基,所述取代芳基为被选自卤素、c1-c6直链或支链的烷基和c1-c2直链或支链的烷氧基所取代的苯基;
在上述实施例中,更为优选地方案为:所述取代芳基为被选自f、cl、br、甲基、乙基、正丙基、叔丁基、正戊基、甲氧基、乙氧基所取代的苯基
在一些实施例中,所述r为芳杂基,所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;所述直链烷基选自c3-c7直链烷基
在上述实施例中,更为优选地方案为:所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;
在一些实施例中,所述r为直链烷基,所述直链烷基选自辛基,庚基,己基。
在一些实施例中,所述三氟苯乙酮化合物具有式(iii)所示结构:
其中,基团r1选自芳基、取代芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基。
在一些实施例中,所述基团r1为芳基,所述芳基为苯基;
在一些实施例中,所述基团r1为取代芳基,所述取代芳基选自br、甲基、甲氧基、硝基所取代的苯基;
在一些实施例中,所述基团r1为芳杂基,所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶。
在一些实施例中,所述“将烯基叠氮、三氟苯乙酮混合均匀”的具体方法为:将式(ii)化合物、式(iii)化合物加入到溶剂中加热溶解;
在一些实施例中,所述合成过程中,体系中还加入五甲基二乙烯三胺(pmdeta)、三苯基膦。
在一些实施例中,所述溶剂选自水、1,2-二氯乙烷、乙二醇、甲醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺(dmf)、乙二醇、二甲基亚砜(dmso)中的一种或多种,
在一些实施例中,所述反应温度为0-120℃,最优选为40℃;
在一些实施例中,所述式(ii)、式(iii)、五甲基二乙烯三胺、三苯基膦的摩尔比为1:(1-2):(1-10):(1-10),最佳反应摩尔比为1:1:2:1;
在一些实施例中,所述醋酸铜加入量为烯基叠氮的1%-100%,最优值为15%,所述百分比为摩尔百分比;
在一些实施例中,加入催化剂后,继续加热搅拌0-24h,优选为6h。
在一些实施例中,本申请的合成方法还包括以下步骤:反应结束后,将反应液冷却后倒入水中,萃取,合并有机相,干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析即得式(i)化合物;
优选地,所述萃取溶剂选自水、1,2-二氯乙烷、乙二醇、甲醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺、乙二醇、二甲基亚砜乙酸乙酯和二氯甲烷,优选为二氯甲烷;
在上述实施例中,更为优选地方案为:所述萃取进行1-3次,每次使用5-20ml萃取溶剂;
在上述实施例中,更为优选地方案为:所述干燥使用无水硫酸镁;
在上述实施例中,更为优选地方案为:所述硅胶柱层析使用的洗脱液为石油醚和乙酸乙酯;
在上述实施例中,更为优选地方案为:所述石油醚与乙酸乙酯的体积比为1-100:1,最佳体积比为60:1。
本发明的有益效果在于:
(1)本发明从简便易得的底物出发,在温和条件下实现快速、高选择性的合成β-三氟甲基-β-羟基-1,2-二苯基丙酮的制备,解决了本领域长期存在的β-三氟甲基-β-羟基-1,2-二苯基丙酮合成条件复杂、选择性差、产率较低等问题,具备广泛的工业应用前景。
(2)本发明的方法简便、高效,所用到的原料简单易得且无毒,步骤少,成本低,纯度高,收率高,适宜大规模的工业化生产。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。
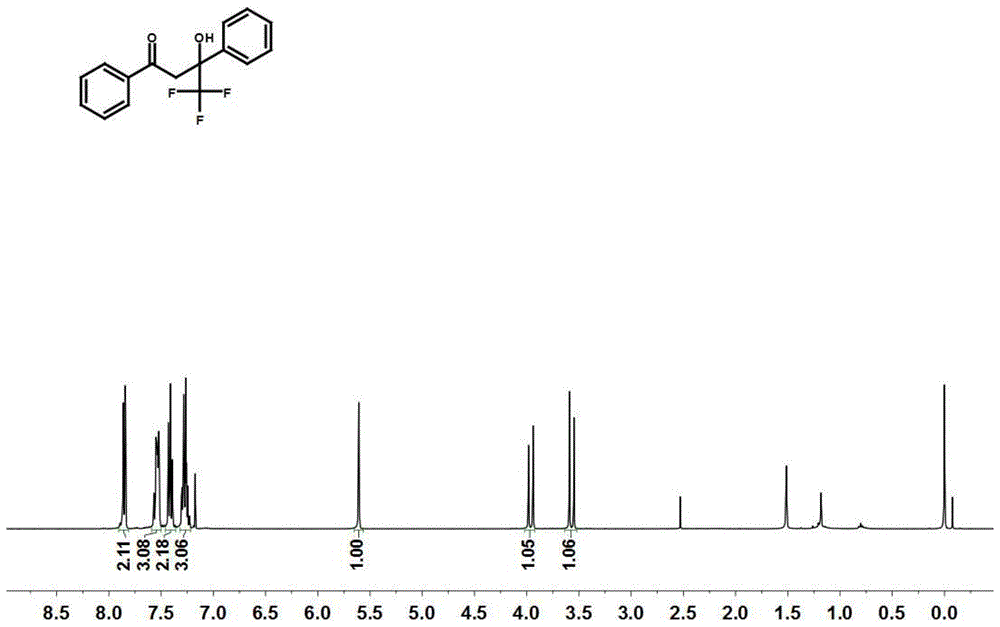
图1为化合物3a的1h-nmr的核磁共振谱;
图2为化合物3a的13c-nmr的核磁共振谱;
图3为化合物3a的19f-nmr的核磁共振谱;
图4为化合物3b的1h-nmr的核磁共振谱;
图5为化合物3b的13c-nmr的核磁共振谱;
图6为化合物3b的19f-nmr的核磁共振谱;
图7为化合物3c的1h-nmr的核磁共振谱;
图8为化合物3c的13c-nmr的核磁共振谱;
图9为化合物3c的19f-nmr的核磁共振谱;
图10为化合物3d的1h-nmr的核磁共振谱;
图11为化合物3d的13c-nmr的核磁共振谱;
图12为化合物3d的19f-nmr的核磁共振谱;
图13为化合物3e的1h-nmr的核磁共振谱;
图14为化合物3e的13c-nmr的核磁共振谱;
图15为化合物3e的19f-nmr的核磁共振谱;
图16为化合物3f的1h-nmr的核磁共振谱;
图17为化合物3f的13c-nmr的核磁共振谱;
图18为化合物3f的19f-nmr的核磁共振谱。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本申请提供进一步的说明。除非另有指明,本申请使用的所有技术和科学术语具有与本申请所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
正如背景技术所介绍的,针对目前β-三氟甲基-β-羟基-1,2-二苯基丙酮合成条件繁琐,产物单一,选择性差、产率较低的问题。因此,本发明提出一种合成式(i)所示化合物的方法,所述方法包括以烯基叠氮、三氟苯乙酮为原料在醋酸铜的催化下一步合成式(i)化合物;所述式(i)的结构式为:
其中,基团r选自富电子或缺电子的芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基;基团r1选自富电子或缺电子的芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基。
优选地,基团r选自芳基、取代芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基;
优选地,所述芳基为苯基;所述取代芳基为被选自卤素、c1-c6直链或支链的烷基和c1-c2直链的烷氧基所取代的苯基;所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;所述直链烷基选自c3-c7直链烷基;
优选地,所述芳基为苯基;所述取代芳基为被选自f、cl、br、甲基、乙基、正丙基、叔丁基、正戊基、甲氧基、乙氧基所取代的苯基;所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;直链烷基选自辛基,庚基,己基;
优选地,基团r1选自芳基、取代芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基;
优选地,所述芳基为苯基;所述取代芳基为被选自卤素、烷基和烷氧基所取代的苯基;所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;
优选地,所述芳基为苯基;所述取代芳基为被选自br、甲基、甲氧基、硝基所取代的苯基;所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;
优选地,所述烯基叠氮的化合物具有式(ii)所示结构:
其中,基团r选自富电子或缺电子的芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基。
优选地,所述芳基为苯基;所述取代芳基为被选自卤素、c1-c6直链或支链的烷基和c1-c2直链或支链的烷氧基所取代的苯基;所述芳杂基中含有一个或多个杂原子,其中杂原子选自n、o和s;所述直链烷基选自c3-c7直链烷基;
优选地,所述芳基为苯基;所述取代芳基为被选自f、cl、br、甲基、乙基、正丙基、叔丁基、正戊基、甲氧基、乙氧基所取代的苯基;所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;直链烷基选自辛基,庚基,己基;
优选地,所述三氟苯乙酮化合物具有式(iii)所示结构:
其中,基团r1选自芳基、取代芳基、芳杂基、稠芳基、直链烷基、环状烷基、含杂原子烷基;
优选地,所述芳基为苯基;所述取代芳基选自br、甲基、甲氧基、硝基所取代的苯基;所述芳杂基选自噻吩、呋喃、吲哚、吡咯、吡啶;
优选地,所述方法按如下所示的反应路线进行:
优选地,上述所述合成方法的步骤包括将式(ii)化合物、式(iii)化合物加入到溶剂中加热溶解,随后向体系中加入五甲基二乙烯三胺(pmdeta)、三苯基膦及醋酸铜催化剂,继续加热搅拌,tlc检测反应。
进一步优选地,所述溶剂选自水、1,2-二氯乙烷、乙二醇、甲醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺(dmf)、乙二醇、二甲基亚砜(dmso)中的一种或多种;优选为1,2-二氯乙烷、乙二醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺(dmf)或二甲基亚砜(dmso),优选为二甲基亚砜(dmso)。提高原料的转化率,同时提高产物的产率。
进一步优选地,所述反应温度为0-120℃,优选为40℃。提高原料的转化率,同时提高产物的产率。
进一步优选地,所述式(ii)、式(iii)、五甲基二乙烯三胺(pmdeta)、三苯基膦的摩尔比为1:(1-2):(1-10):(1-10),最佳反应摩尔比为1:1:2:1;
进一步优选地,所述醋酸铜加入量为1%-100%,优选为15%;
进一步优选地,加入催化剂后,继续加热搅拌0-24h,优选为6h。
优选地,上述所述合成方法还包括以下步骤:
在tlc检测反应结束后,将反应液冷却后倒入水中,萃取,合并有机相,干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析即得式(i)化合物;
进一步优选地,所述萃取溶剂选自水、1,2-二氯乙烷、乙二醇、甲醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺(dmf)、乙二醇、二甲基亚砜(dmso)乙酸乙酯和二氯甲烷;优选为二氯甲烷;
进一步优选地,所述萃取进行1-3次,每次使用5-20ml萃取溶剂;优选地,所述干燥使用无水硫酸镁;
进一步优选地,所述硅胶柱层析使用的洗脱液为石油醚和乙酸乙酯;
进一步优选地,所述石油醚与乙酸乙酯的体积比为(1-100):1,最佳体积比为60:1。
实施例1
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在70℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为8%。
实施例2
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在40℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为90%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
不同溶剂条件下实施例
实施例3
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldce中在40℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为58%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
实施例4
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1ml乙腈中在40℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为70%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
实施例5
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在40℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为90%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
不同催化剂条件下实施例
实施例6
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在40℃条件下加热溶解,随后向体系中加入cubr(0.011g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率61%。
实施例7
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在40℃条件下加热溶解,随后向体系中加入nicl2(0.010g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为67%。
实施例8
将化合物1a即苯基烯基叠氮(0.072g,0.5mmol)、化合物2a即三氟苯乙酮(0.070ml,0.5mmol)、pmdeta(0.208ml,1.0mmol)、三苯基膦(0.130g,0.5mmol)加入到1mldmso中在40℃条件下加热溶解,随后向体系中加入cu(oac)2(0.014g,0.075mmol),继续加热搅拌6h,tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=60:1)得到化合物3a的产率为90%。
反应如下图所示:
化合物3a:
1hnmr(400mhz,cdcl3)δ7.99–7.90(m,2h),7.68–7.59(m,3h),7.55–7.43(m,2h),7.40–7.31(m,3h),5.69(s,1h),4.05(d,j=17.4hz,1h),3.66(d,j=17.4hz,1h);13cnmr(101mhz,cdcl3)δ199.71,137.69,136.32,134.46,128.96,128.76,128.46,128.25,126.32,124.62(d,j=284.8hz),76.39(t,j=28.8hz),40.24;19fnmr(376mhz,cdcl3)δ-80.20;hrms(esi)m/zcalculatedforc16h13f3o2[m+h]+:295.0947,found:295.0939.
-----------------------------------------------------------------------------
实施例9
用4-甲基苯基烯基叠氮(即化合物1b)代替实施例2中的化合物1a,其他条件同实施例2,进行如下所示的反应,得到化合物3b产率为95%。,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%
化合物3b:
1hnmr(400mhz,cdcl3)δ7.87–7.79(m,2h),7.65–7.56(m,2h),7.40–7.26(m,5h),5.80(s,1h),4.00(d,j=17.2hz,1h),3.60(d,j=17.2hz,1h),2.42(s,3h);13cnmr(101mhz,cdcl3)δ199.30,145.67,137.79,133.88,129.64,128.70,128.41,126.33,124.64(d,j=284.7hz),76.82(d,j=84.02hz),39.93,21.77;19fnmr(376mhz,cdcl3)δ-80.18;hrms(esi)m/zcalculatedforc17h15f3o2[m+h]+:309.1103,found:309.1098.
-----------------------------------------------------------------------------------
实施例10
用4-溴苯基烯基叠氮(即化合物1c)代替实施例2中的化合物1a,其他条件同实施例2进行如下所示的反应,得到化合物3c产率为90%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
化合物3c:
1hnmr(400mhz,cdcl3)δ7.83–7.73(m,2h),7.66–7.60(m,2h),7.62–7.55(m,2h),7.40–7.29(m,3h),5.51(s,1h),3.98(d,j=17.3hz,1h),3.61(d,j=17.4hz,1h);13cnmr(101mhz,cdcl3)δ198.59,137.48,135.00,132.34,129.96,129.67,128.85,128.51,126.24,124.53(d,j=284.9hz),76.48(d,j=29.1hz),40.32;19fnmr(376mhz,cdcl3)δ-80.18;hrms(esi)m/zcalculatedforc16h12brf3o2[m+h]+:373.0052,found:373.0045.
---------------------------------------------------------------------------------
实施例11
用3-噻吩烯基叠氮(即化合物1d)代替实施例2中的化合物1a,其他条件同实施例2进行如下所示的反应,高区域选择性得到化合物3d产率为75%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
化合物3d:
1hnmr(400mhz,cdcl3)δ8.18–8.14(m,1h),7.64–7.58(m,2h),7.52–7.48(m,1h),7.43–7.29(m,4h),5.71(s,1h),3.87(d,j=17.0hz,1h),3.59(d,j=17.0hz,1h);13cnmr(101mhz,cdcl3)δ193.42,141.63,137.60,133.90,128.77,128.45,127.19,126.63,126.28,124.56(d,j=284.8hz),76.45(d,j=29.1hz),41.51;19fnmr(376mhz,cdcl3)δ-80.16;hrms(esi)m/zcalculatedforc14h11f3o2s[m+h]+:301.0511,found:301.0470.
----------------------------------------------------------------------------------------
实施例12
用4'-溴-2,2,2-三氟苯乙酮(即化合物2b)代替实施例2中的化合物2a,其他条件同实施例2进行如下所示的反应,高区域选择性得到化合物3e产率为85%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
化合物3e:
1hnmr(400mhz,cdcl3)δ7.98–7.89(m,2h),7.69–7.61(m,1h),7.55–7.43(m,6h),5.70(s,1h),4.01(d,j=17.4hz,1h),3.63(d,j=17.4hz,1h);13cnmr(101mhz,cdcl3)δ199.51,136.83,136.08,134.69,131.68,129.05,128.27,128.18,124.29(d,j=284.6hz),123.24,76.34(d,j=29.2hz),40.00;19fnmr(376mhz,cdcl3)δ-80.34;hrms(esi)m/zcalculatedforc16h12brf3o2[m+h]+:373.0052,found:373.0026.
-----------------------------------------------------------------------
实施例13
用2,2,2-三氟-2,3,4-甲氧基-苯乙酮(即化合物2c)代替实施例2中的化合物2a,其他条件同实施例2进行如下所示的反应,得到化合物3f产率为92%,经过柱层析纯化之后核磁谱图未见明显杂质峰,纯度高于95%。
化合物3f:
1hnmr(400mhz,cdcl3)δ7.95(d,j=7.6hz,2h),7.65–7.54(m,1h),7.53–7.40(m,3h),6.71(d,j=9.0hz,1h),5.85(s,1h),4.47(d,j=17.1hz,1h),3.91–3.83(m,6h),3.70(s,3h),3.57(d,j=17.1hz,1h);13cnmr(101mhz,cdcl3)δ199.02,154.20,151.96,141.87,136.70,133.82,128.68,128.27,124.72(d,j=285.2hz),124.12,121.41,106.82,76.45(d,j=29.5hz),61.03,60.47,55.85,41.03;19fnmr(376mhz,cdcl3)δ-80.82;hrms(esi)m/zcalculatedforc19h19f3o5[m+h]+:385.1264,found:385.1241.
-----------------------------------------------------------------------
最后应该说明的是,以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!