一种连续氧化脱氢制备丙烯腈的方法与流程
本发明涉及有机合成技术领域,尤其涉及一种连续氧化脱氢制备丙烯腈的方法。
背景技术:
丙烯腈是合成纤维、合成橡胶和合成树脂的重要单体。由丙烯腈制得的聚丙烯腈纤维即腈纶,其性能极似羊毛,因此也叫合成羊毛。丙烯腈与丁二烯共聚可制得丁腈橡胶,具有良好的耐油性,耐寒性,耐磨性,和电绝缘性能,并且在大多数化学溶剂、阳光和热作用下,性能比较稳定。丙烯腈与丁二烯、苯乙烯共聚制得的abs树脂,具有质轻、耐寒、抗冲击性能较好等优点。丙烯腈水解可制得丙烯酰胺和丙烯酸及其酯类,它们是重要的有机化工原料。丙烯腈还可电解加氢偶联制得己二腈,由己二腈加氢又可制得己二胺,己二胺是尼龙66原料,可制造抗水剂和胶粘剂等,也用于其他有机合成和医药工业中,并用作谷类熏蒸剂等。此外,该品也是一种非质子型极性溶剂,作为油田泥浆助剂pac142原料。丙烯腈还是制备杀虫剂虫满腈的中间体。
丙烯腈的工业生产方法主要有三种,分别为氰乙醇法、乙炔法、丙烯氨氧化法。
其中,氰乙醇法主要是以环氧乙烷和氢氰酸为原料,在水和三甲胺的存在下反应得氰乙醇,然后以碳酸镁为催化剂,于200-280℃脱水制得丙烯腈,收率约75%。此法生产的丙烯腈纯度较高,但氢氰酸毒性大,成本较高。
乙炔法是以乙炔和氢氰酸为主原料,在氯化亚铜-氯化钾-氯化钠稀盐酸溶液的催化作用下,在80-90℃反应得丙烯腈。此法生产过程简单,收率良好,以氢氰酸计可达97%。但副反应多,产物精制较难,毒性也大,且原料乙炔价格高于丙烯,在技术和经济上落后于丙烯氨氧化法。
丙烯氨氧化法是以丙烯、氨、空气和水为原料,按一定配比进入沸腾床或固定床反应器,在以硅胶作载体的磷钼铋系或锑铁系催化剂作用下,在400-500℃温度和常压下,生成丙烯腈。然后经中和塔用稀硫酸除去未反应的氨,再经吸收塔用水吸收丙烯腈等气体,形成水溶液,使该水溶液经萃取塔分离乙腈,在脱氢氰酸塔除去氢氰酸,经脱水、精馏而得丙烯腈产品,副产品有乙腈、氢氰酸和硫酸铵。此法能耗较大,污染大。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种连续氧化脱氢制备丙烯腈的方法,以丙腈为原料,采用氧化脱氢的新方法制备丙烯腈,工艺路线简单,同时具有较高的收率和纯度。
为解决以上技术问题,本发明提供了一种连续氧化脱氢制备丙烯腈的方法,包括以下步骤:
以丙腈为原料,在镍系催化剂的催化作用下,发生氧化脱氢反应,得到丙烯腈;
所述镍系催化剂由ni,co,mo和zr的氧化物组成。
本发明以丙腈为原料,本发明对所述丙腈的来源并无特殊限定,可以为一般市售,如一般的工业级原料,在本发明的一些具体实施例中,所述丙腈的质量含量为99.5%。
所述镍系催化剂由ni,co,mo和zr的氧化物组成。
本发明优选的,所述ni,co,mo和zr的摩尔比为10~12:3~4:5:2~3。
进一步优选的,所述ni,co,mo和zr的摩尔比为11~12:3~3.5:5:2~2.5。在本发明的一些具体实施例中,所述ni,co,mo和zr的摩尔比为12:3:5:2。
本发明优选的,所述催化剂按照以下方法制备:
将含镍水溶性无机盐、含钴水溶性无机盐、含钼水溶性无机盐、含锆水溶性无机盐在水中混合、溶解,得到混合溶液;
将混合溶液加热反应,冷却、干燥得到催化剂。
本发明首先将含镍水溶性无机盐、含钴水溶性无机盐、含钼水溶性无机盐、含锆水溶性无机盐在水中混合,搅拌溶解,更优选为先将含镍水溶性无机盐、含钴水溶性无机盐、含钼水溶性无机盐混合,调节ph值至7,再与含锆水溶性无机盐混合,搅拌。本发明对于所述搅拌的方式不进行限定,本领域技术人员熟知的即可;所述搅拌的时间优选为20~30min;再加入氢氧化锆的搅拌时间优选为40~60min。
按照本发明,所述含镍水溶性无机盐包括但不限于六水合硝酸镍;所述镍水溶性无机盐质量浓度优选为≥99%。
所述含钴水溶性无机盐包括但不限于六水合硝酸钴;所述含钴水溶性无机盐的浓度优选为0.50~0.75mol/l;所述含钼水溶性无机盐包括但不限于钼酸铵;所述钼水溶性无机盐的质量浓度优选为≥98%。
所述含锆水溶性无机盐包括但不限于氢氧化锆。所述含锆水溶性无机盐的质量浓度优选为≥99.9%。
将混合溶液加热反应,冷却、干燥得到催化剂。
本发明优选将混合溶液转移至聚四氟乙烯反应釜中;本发明优选将反应釜放入160℃的鼓风干燥箱内恒12h。
本发明对于所述冷却不进行限定,自然冷却至室温即可。
冷却后,使用真空泵,抽滤瓶及滤纸抽真空抽滤,使用无水乙醇反复洗涤有机溶剂后再使用蒸馏水洗涤,干燥得到催化剂;所述干燥温度为70~80℃;所述干燥时间为10~12h。
然后通过催化剂成型机,制备固定床用催化剂。
在丙腈氧化脱氢过程中,由于碳氮三键是强极性共价键,反应过程中会导致远离氰基的碳碳键容易断裂,生成乙腈和甲基,甲基相互结合生成乙烷。本发明选用上述特定催化剂,通过调配催化剂中四种贵金属配比,控制催化剂脱氢能力强弱,从而使远离氰基的碳碳键被氧化脱氢,进而抑制碳碳键的断裂,在催化剂的催化作用下,远离氰基的碳碳键被氧化脱氢,生成丙烯腈。
上述反应的反应方程式如下:
本发明优选的,上述反应可以在固定床反应器等连续化化工生产装置中进行。
优选的,所述反应的工艺流程图如图1所示。
具体的,本发明所述反应的反应系统包括依次连接的预混器,固定床反应器,气液分离装置,连续萃取分离装置和连续精馏装置。
其中,预混器用于丙腈液体和氧气的气液前期的混合吸收,保持气液乳化效果,有利于加快反应进度。
本发明对上述固定床反应器并无特殊限定,可以为本领域技术人员熟知的一般固定床反应器,本发明优选采用金属材质的固定床反应器。
本发明将上述镍系催化剂装填于固定床反应器中。
装填过程优选如下:
在固定床反应器底部填装石英砂,中部填装催化剂,顶部再填装石英砂即可。
本发明优选的,所述镍系催化剂占丙腈质量的0.01%~0.50%。
本发明优选的,在反应之前采用氮气吹扫反应系统,氮气由预混器进入反应系统,吹扫速度和时间可根据需要设置,能够隔绝氧气,即避免氧气或空气进入反应系统即可,本发明对此并无特殊限定。优选的,所述氮气的吹扫速度为10~40ml/min,时间为5~20min。在本发明的一些具体实施例中,吹扫速度为20ml/min,时间为10min。
提前采用氮气对反应系统进行吹扫可以隔绝氧气,在无丙腈通入情况下,保证反应系统在升温过程中,催化剂处于无氧状态,防止催化剂结焦,保证催化剂活性。
同时,将固定床反应器温度由室温升至200~220℃,对固定床反应器中的催化剂进行活化。
所述升温的速度可以根据实验情况自行设置,优选为10~30℃/min。在本发明的一些具体实施例中,所述升温的速度为20℃/min。缓慢升温有利于催化剂的缓慢活化。
本发明中,所述反应以空气或氧气为氧化剂。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气或氧气。优选的,所述空气或氧气流量由0ml/min缓慢增加至1500ml/min。本发明对所述空气或氧气流量的增加速率并无特殊限定,可根据需要或反应情况自行设置,能够稳定气流,并使反应平稳进行即可。优选的,所述空气或氧气流量的增加速度为50~200ml/min。在本发明的一些具体实施例中,所述空气或氧气流量的增加速度为100ml/min。
在通入空气的同时,将氮气流速缓慢降至0ml/min。本发明对所述氮气流量的降低速率并无特殊限定,可根据需要或反应情况自行设置,能够稳定气流,并使反应平稳进行即可。优选的,所述氮气流量的降低速度为5~20ml/min。在本发明的一些具体实施例中,所述氮气流量的降低速度为10ml/min。
本发明优选的,通过缩小出口的开度,在固定床反应器末端缓慢背压至反应适合压强,优选的,背压至1.0~7.0mpa。在本发明的一些具体实施例中,背压至2.0mpa。
当压力稳定后,由预混器开始向反应系统通入液相丙腈,所述液相丙腈和空气在预混器进行气液前期的混合吸收,并平稳进入反应系统。
所述丙腈的流速优选为2~7ml/min,在本发明的一些具体实施例中,所述丙腈的流速为5ml/min。
当丙腈和空气的混合物进入固定床反应器后,在镍系催化剂的作用下,进行氧化脱氢反应,得到丙烯腈。
所述反应的停留时间优选为≤5min。
所述反应的温度优选为200~450℃,反应的压强优选为1.0~7.0mpa。
在这样的压力条件下氧气浓度更高,氧化更充分,产率更高。
所述丙腈和空气的摩尔比优选为1:(5~15);所述丙腈和氧气的摩尔比优选为1:(1~3)。
反应结束后,反应液进入气液分离装置,回收的气体可以回用。
液相物料进入连续萃取分离装置,其中,水相储存备用,油相进入连续精馏装置,经过连续精馏处理后,得到丙烯腈,少量的副产物丙腈可回用。
本发明提供的上述连续氧化脱氢制备丙烯腈的方法,产物中只有未反应完全的丙腈、产物丙烯腈和水,反应产物经分离后得到丙腈和丙烯腈,丙腈可重复利用。该工艺路线简单,清洁无污染,后续分离流程短,对设备无腐蚀,而且产率高,具有较大的工业应用价值。另外,丙腈一般只作为溶剂或者医药中间体,用途较少,市场容量小,而丙烯腈用途广泛,将丙腈转化为丙烯腈后,大大增加了丙腈的市场容量。
附图说明
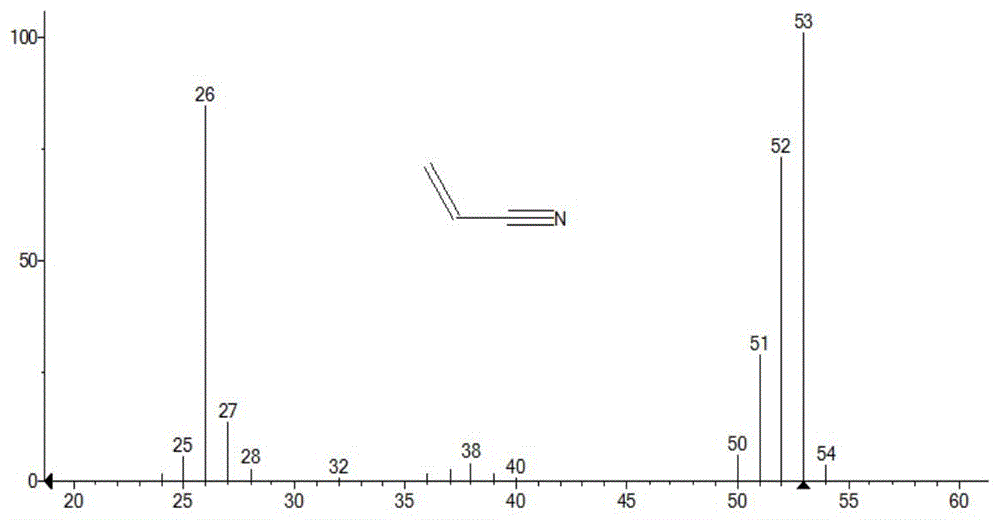
图1为丙烯腈标准质谱图;
图2本发明实施例2反应得到样品质谱图;
图3为本发明提供的连续氧化脱氢制备丙烯腈反应的工艺流程图。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的连续氧化脱氢制备丙烯腈的方法进行详细描述。
以下实施例中,反应器采用管式固定床反应器。
其中,气体回用采取气体压缩机压缩,打入反应系统即可。丙腈通过精馏得到纯品,采用进料泵打入反应系统。气液分离采用气液分离罐进行分离。
实施例1
称取30.00g六水合硝酸镍加至80ml浓度为0.5mol/l的六水合硝酸钴溶液中,搅拌使其溶解,然后称取10.00g钼酸铵,溶于上述溶液中,再将2.5mol/l的naoh溶液加到上述溶液中,调节ph至7,搅拌30min,然后加入7.5g氢氧化锆,继续搅拌1.0h。然后将混合液转入到聚四氟乙烯反应釜中,并将反应釜放入160℃的鼓风干燥箱内恒12h。将反应釜取出,冷却至室温,使用真空泵;抽滤瓶及滤纸抽真空抽滤,使用无水乙醇反复洗涤有机溶剂后再使用蒸馏水洗涤,放入烘箱80℃干燥12小时,得到催化剂样品,然后通过催化剂成型机,制备固定床用催化剂。
实施例2
称取48.40g六水合硝酸镍加至80ml浓度为0.5mol/l的六水合硝酸钴溶液中,搅拌使其溶解,然后称取14.85g钼酸铵,溶于上述溶液中,再将2.5mol/l的naoh溶液加到上述溶液中,调节ph至7,搅拌30min,然后加入11.38g氢氧化锆,继续搅拌1.0h。然后将混合液转入到聚四氟乙烯反应釜中,并将反应釜放入160℃的鼓风干燥箱内恒12h。将反应釜取出,冷却至室温,使用真空泵;抽滤瓶及滤纸抽真空抽滤,使用无水乙醇反复洗涤有机溶剂后再使用蒸馏水洗涤,放入烘箱80℃干燥12小时,得到催化剂样品,然后通过催化剂成型机,制备固定床用催化剂。
实施例3
称取52.87六水合硝酸镍加至80ml浓度为0.5mol/l的六水合硝酸钴溶液中,搅拌使其溶解,然后称取14.85溶于上述溶液中,再将2.5mol/l的naoh溶液加到上述溶液中,调节ph至7,搅拌30min,然后加入7.58氢氧化锆,继续搅拌1.0h。然后将混合液转入到聚四氟乙烯反应釜中,并将反应釜放入
实施例4
在固定床反应器中填装10ml实施例1制备的镍系催化剂(ni,co,mo和zr的摩尔比为10:3:5:3),装填过程为:在固定床底部填装石英砂,中部填装催化剂,顶部再填装石英砂。氮气与预混器连接,从预混器进入,吹扫反应系统,吹扫速度为20ml/min,吹扫时间为10min,并缓慢将反应温度由室温升温至200℃,升温速度为20℃/min。
当反应温度达到预定温度时,从预混器开始向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢降至0ml/min,降低速率为10ml/min。通过缩小出口的开度,在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,从预混器中开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量38.6%,丙烯腈含量60.7%。反应收率56.9%。
采用质谱(mass)对反应产物结构进行表征。图1为丙烯腈标准质谱图,图2本发明实施例2反应得到样品质谱图。通过图1以及图2可以确定,本发明实施例2制备得到的是丙烯腈。
实施例5
在固定床反应器中填装10ml实施例1制备的镍系催化剂(ni,co,mo和zr的摩尔比为10:3:5:3),装填过程同实施例2,氮气以20ml/min吹扫反应系统,吹扫时间为15min,并缓慢将反应温度由室温升温至300℃,升温速度为20℃/min。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢将至0ml/min,降低速率为10ml/min。在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量28.4%,丙烯腈含量70.5%。反应收率66.9%。
实施例6
在固定床反应器中填装20ml实施例1制备的镍系催化剂(ni,co,mo和zr的摩尔比为10.0:3:5:3),装填过程同实施例2,氮气以20ml/min吹扫反应系统,吹扫时间为20min,并缓慢将反应温度由室温升温至300℃,升温速度为20℃/min。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢将至0ml/min,降低速率为10ml/min。在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量15.1%,丙烯腈含量83.8%。反应收率78.5%。
实施例7
在固定床反应器中填装25ml实施例1制备的镍系催化剂(ni,co,mo和zr的摩尔比为10:3:5:3),装填过程同实施例2,氮气以20ml/min吹扫反应系统,吹扫时间为20min,并缓慢将反应温度由室温升温至300℃,升温速度为20℃/min。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢将至0ml/min,降低速率为10ml/min。在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量8.1%,丙烯腈含量90.8%。反应收率86.7%。
实施例8
在固定床反应器中填装20ml实施例2制备的镍系催化剂(ni,co,mo和zr的摩尔比为12:3:5:2),装填过程同实施例2,氮气以20ml/min吹扫反应系统,吹扫时间为20min,并缓慢将反应温度由室温升温至400℃,升温速度为20℃/min。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢将至0ml/min,降低速率为10ml/min。在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量8.1%,丙烯腈含量90.8%。反应收率86.0%。
实施例9
在固定床反应器中填装20ml实施例3制备的镍系催化剂(ni,co,mo和zr的摩尔比为12:4:5:2),装填过程同实施例2,氮气以20ml/min吹扫反应系统,吹扫时间为20min,并缓慢将反应温度由室温升温至400℃,升温速度为20℃/min。
当反应温度达到预定温度时,开始从预混器向反应系统通入空气,空气流量由0ml/min缓慢增加至1500ml/min,空气流量增加的速率为100ml/min,同时,将氮气流速由20ml/min缓慢将至0ml/min,降低速率为10ml/min。在固定床反应器末端缓慢背压至2.0mpa,当压力稳定后,开始通入液相丙腈,丙腈流速为5ml/min,反应停留时间≤5min,稳定出料后,收集反应液,萃取分离后,取油相气相色谱分析检测分析,其中丙腈含量8.1%,丙烯腈含量90.8%。反应收率83.5%。
由上述实施例可知,本发明提供的上述方法,制备得到了丙烯腈,且工艺路线简单,同时具有较高的收率和纯度。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!