FCHO2基因在制备宫颈癌放疗增敏药物的应用的制作方法
本发明属于生物医药领域,具体涉及fcho2基因在制备宫颈癌放疗增敏药物的应用。
背景技术:
肿瘤放疗增敏药物:放射治疗是治疗恶性肿瘤的重要手段之一。放射增敏药物是一种化学或药物制剂,当与放射治疗同时应用时可以改变肿瘤细胞对放射的反应性,从而增加对肿瘤细胞的杀伤效应。放射增敏是指为增强射线对肿瘤细胞的杀伤效应,提高肿瘤的控制率和治愈率。在分子生物学指导下提高肿瘤对射线的敏感性以及改善放射抗拒肿瘤的放射效果是放疗发展新趋势,放疗正从“物理精确”走向“生物精准”。
宫颈癌是我国女性生殖道二大恶性肿瘤之首,尤其是近年来出现全球发病年轻化趋势,严重威胁着广大妇女的健康。其中ⅱb期以上中晚期患者众多,其治疗手段主要采用以放疗为主的综合治疗。放射治疗是中晚期宫颈癌中的主要治疗手段,体外照射联合腔内近距离放疗是公认的放疗模式。接受单纯放疗的ⅱb-ⅳ期患者的5年生存率在50%-70%和30%-50%之间。尽管近年来放疗新技术不断取得进步,放化疗及靶向的新药物为宫颈癌患者带来新的希望,但宫颈癌根治性放疗为主的综合治疗疗效并未有明显改善。
目前同步放化疗为中晚期宫颈癌标准治疗方案,宫颈癌细胞本身基因特性导致其临床效果差异很大,其中宫颈癌细胞的放射敏感性与其细胞分化程度、细胞周期、临床分期和解剖大体分型等诸多因素有关,特别肿瘤的基因表型。随着分子生物学的发展,研究者从分子水平上阐述了宫颈癌的发病机制,发现多个基因分子水平的变化与宫颈癌的发生、发展密切相关,在宫颈癌的放疗反应中起着重要作用。人们试图从基因水平寻找对辐射敏感的放疗靶点,以最高效的杀灭肿瘤,同时最大限度地保护正常组织,从而提高放疗效果,减少放疗副作用。尽管目前研究很多,但临床仍未有高级别循证医学证据的放疗增敏药物。需要进一步筛选与验证。
技术实现要素:
本发明的目的在于提fcho2基因在制备宫颈癌放疗增敏药物的应用。
为实现上述目的,本发明采用如下技术方案:
通过表达谱芯片筛从宫颈癌根治性放疗临床标本中筛选出宫颈鳞癌放疗敏感性基因,进一步通过高通量慢病毒高内涵(hcs)细胞筛选实验方法,通过比较基因敲减放疗处理前后细胞增殖速度的影响,在待检目的基因中初步筛选出放疗后增殖抑制表型明显的fcho2基因。
制备fcho2放射敏感性基因rnai慢病毒的药物,从而提高宫颈癌的放射敏感性,进而提高肿瘤的局控率及生存率。
本发明的优点在于:fcho2基因rnai慢病毒载体的设计、构建,本发明将fcho2基因rnai慢病毒转染宫颈癌细胞后再放疗,能够抑制细胞增殖、细胞周期阻滞及促进细胞凋亡,具有靶向宫颈癌放射敏感性的作用。本发明提供了fcho2基因和抑制或下调fcho2基因表达的试剂的新应用,能够有效提高宫颈癌肿瘤细胞的放疗敏感性。
附图说明
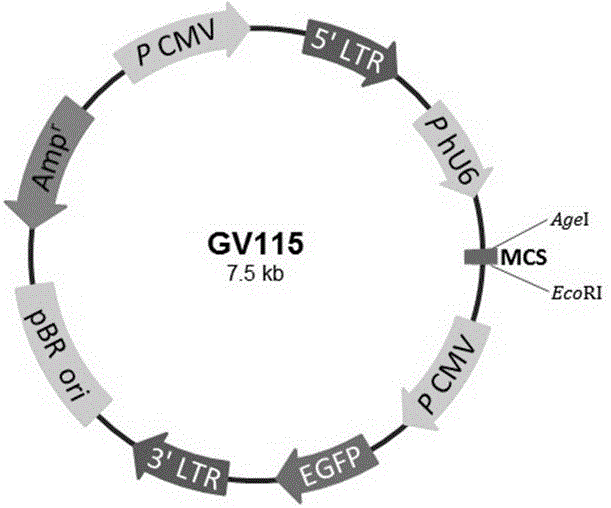
图1工具载体图谱。
图2线性化载体电泳图。其中泳道1:1kbmarker:从上至下依次为10kb,8kb,6kb,5kb,4kb,3.5kb,3kb,2.5kb,2kb,1.5kb,1kb,750bp,500bp,250bp;泳道2:agei和ecori双酶切线性化后的载体质粒;泳道3:没有酶切的载体质粒。
图3pcr扩增电泳图,泳道1:阴性对照(ddh2o),排除系统中外源核酸污染导致假阳性结果,泳道2:自连对照(空载体自连对照组);泳道3:250bpmarker:从上至下依次为5kb,3kb,2kb,1.5kb,1kb,750bp,500bp,250bp,100bp;泳道4-8:单克隆psc56975-1,2,3,4,5。
图4流式细胞仪检测fcho2基因消减对凋亡的影响。相比shctrl-w组,shfcho2-w组凋亡数无显著差异(p>0.05),相比shctrl-f组,shfcho2-f组凋亡数增多(p<0.05)。
图5流式细胞仪检测fcho2基因消减对细胞周期的影响。相比shctrl-w组,shfcho2-w组处于s期、g1期、g2/m期的细胞均无显著变化;相比shctrl-f组,shfcho2-f组处于s期的细胞增多(p<0.05),处于g1期的细胞无显著变化,g2/m期的细胞减少(p<0.05)。
图6mtt检测fcho2基因消减对细胞增殖的影响。相比shctrl-w组,shfcho2-w组细胞增殖无显著差异(p>0.05);相比shctrl-f组,shfcho2-f组细胞增殖减缓(p<0.05)。
图7westernblot检测靶点降低fcho2基因蛋白表达水平;柱形图代表三次实验的平均值,误差线表示标准偏差(sd),**,shctrl与目的基因shrna慢病毒处理组相比,p<0.01;*,shctrl与目的基因shrna慢病毒处理组相比,0.01<p<0.05。
具体实施方式
实施例1
以fcho2基因为模板,设计rna干扰靶点序列,构建目的基因rna干扰慢病毒载体。
1.实验质粒:
载体编号:gv115,
元件顺序:hu6-mcs-cmv-egfp,
对照插入序列:ttctccgaacgtgtcacgt;
工具载体图谱如图1所示。
2.rna干扰靶点设计
根据rna干扰序列设计原则,以fcho2基因为模板,设计多个19-21ntrna干扰靶点序列。经设计软件评估测定后,选取以下序列作为干扰靶点。
3.dnaoligo序列合成
根据已选靶点序列设计shrna干扰序列,并在两端添加合适的限制性内切酶酶切位点以完成载体构建。除此之外,在正链3’端添加ttttt终止信号,而反链5’端添加终止信号互补序列。设计完成后送捷瑞公司合成单链dnaoligo。
*ccgg:agei酶切位点;aattc:ecori酶切位点;g:ecori酶切位点互补序列。
4.fcho2基因rnai慢病毒载体构建和包装:按实验手册进行。
4.1线性化载体制备
按照neb说明书配制50μl反应体系,使用agei和ecori双酶切gv115载体使其线性化。
37℃(最适温度)反应1h,之后切胶回收目的片段。
琼脂糖凝胶电泳图如图2所示。
4.2rna干扰慢病毒载体构建
(1)连接
按照fermentast4dnaligase说明书配制20μl反应体系,将双链dnaoligo与线性化的载体相连。
于16℃反应1h-3h,连接产物命名为psc56975,之后进行转化实验。
(2)转化
将连接产物转化大肠杆菌感受态细胞,详细操作步骤如下:
将10μl连接产物psc56975加入到100μl大肠杆菌感受态细胞中,冰浴30min。
42℃热激90sec,冰浴2min。
加入500µl无抗生素的lb液体培养基,200rpm于37℃摇床震荡培养1hr。
取150μl菌液均匀涂抹在含有amp的lb固体培养基上,于37℃培养箱中过夜培养。
(3)阳性克隆的pcr鉴定
(3.1)引物
(3.2)pcr扩增
按下表配制20μlpcr反应体系,用无菌枪头挑取单个菌落为模板,进行pcr扩增,反应条件为:94℃3min;94℃30s,55℃30s,72℃30s,22次循环;72℃5min。pcr结束后,取5μl产物,1%琼脂糖凝胶电泳检测条带。
琼脂糖凝胶电泳图片如图3所示。
pcr条带大小:连接入shrna片段的阳性克隆pcr片段大小为:341bp;没有连接入shrna片段的空载体克隆pcr片段大小为:307bp。由此判断psc56975-1,3,4,5为阳性克隆,保存鉴定结果正确的克隆,并进行测序。
5.阳性克隆测序结果分析
以鉴定引物-f进行阳性克隆测序,挑选测序结果与目标序列完全一致的克隆用于下一步实验。psc56975测序结果如下:
aatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgggctacagtattaaaccagaaactcgagtttctggtttaatactgtagctttttgaattcggatccattaggcggccgcgtggataaccgtattaccgccatgcattagttattaatagtaatcaattacggggtcattagttcatagcccatatatggagttccgcgttacataacttacggtaaatggcccgcctggctgaccgcccaacgacccccgcccattgacgtcaataatgacgtatgttcccatagtaacgccaatagggactttccattgacgtcaatgggtggagtatttacggtaaactgcccacttggcagtacatcaagtgtatcatatgccaagtac。
*shrna干扰序列插入片段用斜体黑体标注,其中agei酶切位点被破坏。
质粒抽提:将测序正确的菌液转接于150ml含amp抗生素的lb液体培养基中,37℃摇床震荡培养过夜。按照endofreemaxiplasmidkit说明书提取质粒,质检合格的质粒进入下游流程。
具体步骤如下:
1.8000rpm离心4min收集菌体。
2.加入7mlp1,震荡混匀;
3.加入7mlp3,颠倒混匀6~8次,静置5min;
4.加入7mlp4,颠倒混匀6~8次,冰浴10min;
5.9000rpm离心10min,将上清转移到过滤器cs中,过滤后加入10ml异丙醇混匀;
6.向吸附柱中加入2.5ml的平衡液bl,8000rpm离心2min,倒掉收集管中的废液,将柱子放回备用;
7.将上清分两次倒入吸附柱中,8000rpm离心2min,弃废液;
8.向吸附柱中加入10ml漂洗液pw(已加入无水乙醇),相同转速离心2min,弃废液,重复该步骤一次;
10.向吸附柱中加入3ml无水乙醇,8000rpm离心2min,弃废液;
11.9500rpm空甩5min,除去残留的漂洗液;
将吸附柱转移至一新的白管中,在柱中心滴加800μl洗脱缓冲液tb(先预热),室温放置5min,然后9500rpm离心2min;
12.将管中的洗脱液转移到一干净的1.5mlep管中,-20℃保存;
13.取样电泳,使用分光光度计(thermo_nanodrop2000)测定质粒浓度,质检。
14.将质检合格的质粒转交下游平台进行病毒包装。
6.fcho2基因rna干扰序列的慢病毒感染siha宫颈鳞癌细胞
6.1准备目的细胞
(1)从液氮罐中取出细胞冻存管,迅速放入37℃水浴中,并同时摇动使其尽快解冻。
(2)完全解冻后,1300rpm,离心3min,75%酒精擦拭冻存管消毒后,移至生物安全柜。
(3)吸去冻存液上清,加入1ml新鲜的完全培养基重悬细胞,将细胞悬液接种至含有3ml完全培养基的6-cmdish中,轻轻晃匀后置于37℃、5%co2培养箱。
(4)24h后更换一次培养液继续培养,待细胞汇合度达80%左右传代培养,保持细胞良好的生长状态。
6.2目的细胞慢病毒感染
(1)贴壁细胞:
a.处于对数生长期的细胞胰酶消化,完全培养基制成3-5×104个/ml细胞悬液,接种相应的细胞数到培养板中,继续培养保证感染时铺板量达到15-30%左右。
b.按照预实验结果,更换感染培养基,加入最适病毒量进行感染。
c.参照预实验结果,选择感染后最适时间点更换为常规培养基继续培养,为感染后8-12h左右。
(2)悬浮细胞:
a.按照预实验确定的感染条件,使用感染液将细胞制成3-5×105个/ml悬液,接种相应的细胞数到培养板中。
b.铺板后无需培养,参照预实验确定的moi加入最适病毒量感染。
c.参照预实验结果,选择感染后最适时间点,一般为8-12h左右,将各孔中细胞收集到干净的1.5mlep管中,以2000rpm离心2min,去掉上清液,更换为完全培养基,轻轻混匀后继续培养(备注:如48孔板感染可扩大至24孔板继续培养)。
6.3观察感染后细胞状态及感染效率
(1)细胞状态良好,未出现大量死亡现象,特别是保证nc组不con组细胞状态相当。
(2)一般而言,细胞感染效率达到70%以上,就可以继续下游实验。
a.荧光标记的慢病毒感染。感染后72h左右,荧光显微镜观察报告基因(如gfp)的表达情况,荧光率即为阳性感染率;
b.抗性基因标记的慢病毒感染。一般于感染48-72h后,换用含抗生素(如puromycin)的培养基筛选细胞,细胞存活率即为阳性感染率。
6.4细胞状态良好,感染效率合格组,可用于下游检测;否则重新感染。
6.5.siha宫颈鳞癌细胞的照射:下调fcho2基因的siha宫颈鳞癌细胞4gy照射,观察其对细胞功能的影响。实验步骤如下:
6.5.1细胞凋亡检测:
(1)待各实验组6孔板细胞生长至覆盖率约为70%时药物诱导凋亡。
(2)若为贴壁细胞,则上清中细胞也需收集。胰酶消化,完全培养基重悬成细胞悬液,
与上清细胞收集于同一5ml离心管中,每组设三个复孔(为保证上机细胞数足够,细胞数目≥5×105/处理)。若为悬浮细胞,直接收集。
(3)1300rmp离心5min,弃上清,4℃预冷的d-hanks(ph=7.2~7.4)洗涤细胞沉淀。
(4)1×bindingbuffer洗涤细胞沉淀一次,1300rmp、3min离心,收集细胞。
(5)200μl1×bindingbuffer重悬细胞沉淀。
(6)加入10μlannexinv-apc染色,室温避光10-15min。
(7)根据细胞量,补加400-800μl1×bindingbuffer,上机检测。
6.5.2细胞周期检测
(1)若为贴壁细胞,待各实验组6cmdish细胞生长至覆盖率约为80%时(细胞未进入生长平台期),胰酶消化,完全培养基重悬成细胞悬液,收集细胞于5ml
离心管中,每组设三个复孔(为保证上机细胞数足够,细胞数目≥106/处理)。若为悬浮细胞,直接收集。
(2)1300rmp离心5min,弃上清,4℃预冷的d-hanks(ph=7.2~7.4)洗涤细胞沉淀1次。
(3)1300rmp、5min离心,4℃预冷的75%乙醇固定细胞至少1h。
(4)1300rmp离心5min去固定液,d-hanks洗涤细胞沉淀一次,同步骤(2)。
(5)细胞染色液配制:40×pi母液(2mg/ml):100×rnase母液(10mg/ml):1×d-hanks=25:10:1000。
(6)细胞染色:根据细胞量,加入一定体积的细胞染色液(0.6-1ml)重悬,使上机时细胞通过率为300~800cell/s。
(7)上机检测。
6.5.3细胞增殖活力检测
(1)将处于对数生长期的各实验组细胞胰酶消化后,完全培养基重悬成细胞悬液,并计数。
(2)根据细胞生长快慢决定铺板细胞密度(多数为2000cell/well),每组3-5重复,根据实验设计决定铺板数量(如检测5天,则铺5张96孔板)。
(3)统一铺好后,待细胞完全沉淀下来后,在显微镜下观察各实验组的细胞密度,如果密度不均匀,则固定一组,微调其他组细胞的量再次铺板(如:发现con组细胞较多,降低细胞量再次铺板),放入细胞培养箱中培养。
(4)从铺板后第二天开始,培养终止前4h加入20μl5mg/ml的mtt于孔中,无需换液。
(5)4h后完全吸去培养液,注意不要吸掉孔板底部的甲瓒颗粒,加100μldmso溶解甲瓒颗粒。
(6)振荡器振荡2-5min,酶标仪490/570nm检测od值。
7.fcho2在siha宫颈癌细胞放疗中的细胞功能检测
通过mtt、流式细胞仪检测方法,证实干扰目的基因fcho2对siha宫颈癌细胞放疗后增殖有明显的抑制作用,见图4-7。由图4可以得出,hrna慢病毒感染后第3天放疗,第3天检测,放疗前实验组发生凋亡的siha细胞无显著变化,提示放疗前fcho2基因与siha细胞的凋亡不相关;放疗后实验组发生凋亡的siha细胞显著增加,提示放疗后fcho2基因与siha细胞的凋亡显著相关。图5中,shrna慢病毒感染后第3天放疗,第3天检测,放疗前实验组处于g1、s、g2/m期的细胞均无显著变化,提示放疗前fcho2基因与siha细胞的周期不相关;放疗后实验组处于s期的细胞增多,处于g1期的细胞无显著变化,g2/m期的细胞减少,提示放疗后fcho2基因与siha细胞的周期分布显著相关。图6中,经shrna慢病毒感染后,细胞铺于96孔板,铺板数为2000。连续检测5天,发现放疗前实验组siha细胞的增殖速率无显著变化,提示fcho2基因与siha细胞的增殖能力不相关;放疗后实验组siha细胞的增殖速率受到显著抑制,提示放疗后fcho2基因与siha细胞的增殖能力显著相关。图7中,从westernblot结果可以看出,在293t细胞中,靶点对fcho2基因的外源表达在蛋白质水平有显著的敲减作用。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
sequencelisting
<110>中国人民解放军南京军区福州总医院
<120>fcho2基因在制备宫颈癌放疗增敏药物的应用
<130>5
<160>5
<170>patentinversion3.3
<210>1
<211>19
<212>dna
<213>人工序列
<400>1
ttctccgaacgtgtcacgt19
<210>2
<211>19
<212>dna
<213>人工序列
<400>2
tacagtattaaaccagaaa19
<210>3
<211>18
<212>dna
<213>人工序列
<400>3
cctatttcccatgattcc18
<210>4
<211>17
<212>dna
<213>人工序列
<400>4
gtaatacggttatccac17
<210>5
<211>475
<212>dna
<213>人工序列
<400>5
aatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgctta60
ccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaaca120
ccgggctacagtattaaaccagaaactcgagtttctggtttaatactgtagctttttgaa180
ttcggatccattaggcggccgcgtggataaccgtattaccgccatgcattagttattaat240
agtaatcaattacggggtcattagttcatagcccatatatggagttccgcgttacataac300
ttacggtaaatggcccgcctggctgaccgcccaacgacccccgcccattgacgtcaataa360
tgacgtatgttcccatagtaacgccaatagggactttccattgacgtcaatgggtggagt420
atttacggtaaactgcccacttggcagtacatcaagtgtatcatatgccaagtac475
- 还没有人留言评论。精彩留言会获得点赞!