多拉菌素晶型A、晶型B及其制备方法与流程
本发明属于兽药、化学工程结晶技术领域,具体涉及多拉菌素晶型A、晶型B及其制备方法。
背景技术:
多拉菌素为20世纪90年代由美国辉瑞公司研制开发出的新一代大环内酯类抗寄生虫药,被认为是目前阿维菌素类药物中最优秀的药物之一。多拉菌素是由基因重组的阿维链霉菌(Streptomyces avermitilis)新菌株通过添加环已甲酸为前体物质发酵而成的一种十六元大环内酯类半合成抗生素,属于第三代阿维菌类药物,易溶于有机溶剂,比如甲醇、乙醇、丙酮、1,2-丙二醇、乙酸乙酯、二甲基亚矾、乙酸异丙酯中,其结构式如下所示:
多拉菌素为新型、广谱抗寄生虫药,对胃肠道线虫、肺线虫、螨、蜱和伤口蛆等均有效,用于治疗家畜线虫病和螨病等体外寄生虫病,主要适用于牛和猪。
医药或兽医学化合物的不同固态形式决定了其具有不同的物理性质。物理性质的差异对医药或兽医学化合物的制备、加工、配制或应用都会产生影响。晶型不同会造成其稳定性、溶解度、溶出速率、堆密度、流动性、悬浮稳定性、研磨期间的稳定性、蒸气压力、光学和机械性质、吸湿性、晶体尺寸、过滤性质、干燥、密度、熔点、降解稳定性、防止转变为其它晶型的稳定性等物理化学性质产生重要影响,从而可能会造成其在使用过程中产生不同的药效和生物利用度。因此,对固体药品而言,不同固体形态的开发更有利于根据药物制剂特点选择合适的晶型。
目前,市售多拉菌素主要以外用的浇泼剂及内用的注射剂两种剂型为主,这两种剂型均具有局限性。浇泼剂为体表靶向制剂,由于给药方式的局限性,生物利用度较低;多拉菌素注射剂虽具有良好的杀虫效果,但易对病畜造成较大的应激性,不利于规模化给药。所以,目前急需开发多拉菌素产品的新剂型,而新剂型的开发更离不开稳定晶型的开发,以此来进一步满足兽药市场需求。
现有技术公开了多拉菌素的几种纯化方法,如中国专利CN 109651465 A公开了结晶过程中使用的有机溶剂为乙酸乙酯、乙醇、丙酮,优选乙酸乙酯;但这些溶剂溶解度较大,使得导致精制收率较低。专利CN 108976270 A公开了一种纯化方法,采用丙酮/水重结晶,由于丙酮溶解度较大,加入水量较少,收率在70%左右,造成精制收率偏低,生产成本高。专利CN104693254A公开了一种多拉菌素的结晶方法,得到高纯度的多拉菌素。重结晶溶剂为甲醇、乙醇、丙酮、异丁醇、异丙醇或其中任意的混合溶剂。同样的,由于重结晶溶剂溶解度较好,使得精制收率较低。专利CN 104418927 A公开了一种纯化方法,结晶溶剂为醇/水混合溶剂。所述的醇的水溶液为醇含量在90%以上(V/V),优选95%以上,用量为滤饼重量的5~10倍(V/W)。由于醇占比例较大,使得精制收率较低。所有专利均没有涉及晶型的研究,使得该品种在兽药上的使用受到很大限制。
技术实现要素:
本发明的目的在于提供多拉菌素多晶型及其制备方法,所得晶型A和B稳定性好、纯度高、流动性好;该制备工艺得到的结晶收率好,适合工业化生产,不仅克服了现有技术存在的缺陷,也为多拉菌素在新剂型开发上提供了更多选择。
本发明的第一方面,提供了两种多拉菌素晶型,晶型A和晶型B。
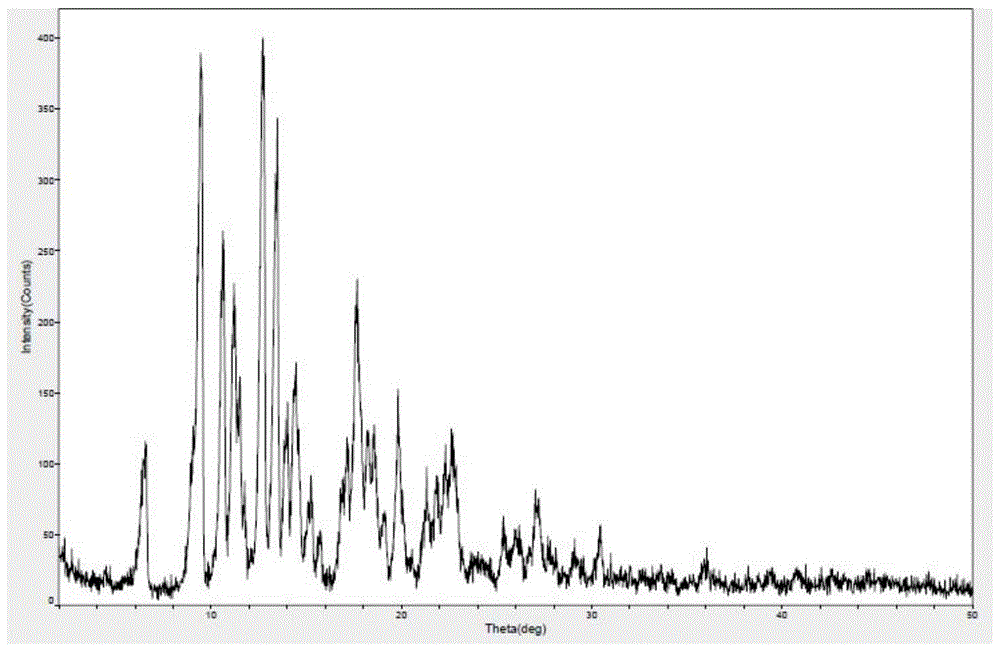
所述晶型A的X射线粉末衍射图(图1)在2θ为6.5±0.2°、9.4±0.2°、10.5±0.2°、11.2±0.2°、 11.5±0.2°、12.7±0.2°、13.4±0.2°、13.9±0.2°、14.4±0.2°、15.2±0.2°、15.7±0.2°、16.7±0.2°、 17.1±0.2°、17.6±0.2°、18.2±0.2°、18.5±0.2°、19.1±0.2°、19.8±0.2°、21.3±0.2°、21.8±0.2°、 22.3±0.2°、22.7±0.2°、25.3±0.2°、27.2±0.2°、30.4±0.2°处有专属特征吸收峰。
所述晶型B的X射线粉末衍射图(图2)在2θ为4.4±0.2°、6.1±0.2°、6.3±0.2°、8.7±0.2°、 9.3±0.2°、10.4±0.2°、10.7±0.2°、11.0±0.2°、11.2±0.2°、11.5±0.2°、12.5±0.2°、13.3±0.2°、 13.8±0.2°、14.2±0.2°、14.4±0.2°、16.9±0.2°、17.6±0.2°、18.0±0.2°、18.2±0.2°、18.5±0.2°、18.9±0.2°、19.8±0.2°、21.4±0.2°、22.0±0.2°、22.5±0.2°、26.8±0.2°、28.1±0.2°、33.0±0.2°、 45.1±0.2°处有专属特征吸收峰。
优选地,本发明提供的多拉菌素晶型A具有的X-射线粉末衍射图如图1所示。
优选地,本发明提供的多拉菌素晶型B具有的X-射线粉末衍射图如图2所示。
另一方面,本发明还提供了多拉菌素晶型A和晶型B的制备方法,其步骤如下:
(1)将多拉菌素置于良溶剂中,热溶解,过滤;
(2)滤液通过减压浓缩并加入不良溶剂置换出良溶剂,析出晶体,留一定体积,降温继续析晶后过滤干燥,得到多拉菌素晶体物。
所用结晶设备选用本领域具有搅拌效果的常规结晶釜即可。
下面对各步进一步说明:
步骤(1)所述良溶剂选自卤代烃类、醇类、酯类、酮类或四氢呋喃等有机溶剂中的一种或几种。
优选地,步骤(1)所述良溶剂选自四氢呋喃、二氯甲烷、氯仿、乙酸乙酯、丙酮、异丙醇、正丁醇、甲醇或乙醇中的一种或几种。
步骤(1)所述多拉菌素与良溶剂用量比(m:v)为1:3-5。
步骤(2)减压浓缩至剩余药液体积为多拉菌素固体重量的1-5倍,优选为2-3倍;加入不良溶剂体积为多拉菌素固体重量的1-5倍,优选为1-2倍。
步骤(2)所述制备晶型A的不良溶剂选自脂肪族烃类溶剂、芳香族烃类、醚类溶剂的一种或几种。
优选地,步骤(2)所述制备晶型A的不良溶剂选自正己烷、环己烷、正庚烷、乙醚、二甲基醚、二异丙基醚、石油醚、甲基叔丁基醚、甲苯中的一种或几种。
更优选地,步骤(2)所述制备晶型A的不良溶剂选自正己烷、正庚烷、石油醚、甲基叔丁基醚、甲苯、乙腈中的一种或几种。
步骤(2)所述制备晶型B的不良溶剂为乙腈。
步骤(1)所述热溶解温度为20-60℃;
步骤(2)所述降温范围为-5-15℃,烘干干燥温度为30-80℃。
有益效果:
1、本发明的精制工艺简单、易操作,适合工业化生产。使用该精制工艺得到的多拉菌素晶体纯度均在99%以上,甚至达到99.5%以上,大大提高了药物的药效;
2、使用该精制工艺得到的多拉菌素晶体的收率均在95%以上,极大的降低了生产成本;
3、该结晶工艺得到的多晶型稳定性好,不会发生晶型转变、不存在吸湿现象。
附图说明
图1是本发明实施例1制备的多拉菌素晶型A的X射线粉末衍射图;
图2是本发明实施例2制备的多拉菌素晶型B的X射线粉末衍射图;
图3是本发明实施例3制备的多拉菌素晶型A的X射线粉末衍射图;
图4是本发明制备的多拉菌素晶体实施例1的HPLC图谱;
图5是本发明制备的多拉菌素晶体实施例2的HPLC图谱;
图6是本发明实施例的原料多拉菌素的HPLC图谱。
具体实施方式
为了对本发明进行进一步的说明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
通用测试方法:
X-射线粉末衍射(XRD)仪器:日本Rigaku D/Max-2500型:辐射源:铜靶在室温条件下扫描:扫描范围:2.0~50.0°,扫描速率:8°/min,步长:0.02°;
纯度检查采用多拉菌素兽药典方法:COSMOSIL C18色谱柱(4.6mm╳150mm,5um);流动相:甲醇:乙腈:水=67:15:18(v/v);波长:245nm;流速:1ml/min;进样量:20ul。
实施例1
取多拉菌素10g,加入四氢呋喃40ml,升温至40℃溶解,减压浓缩至2倍体积,加入 20ml正己烷,继续浓缩至2倍体积,再加入20ml正己烷,继续浓缩至约1倍体积,降温至 0-5℃,过滤,滤饼于40-50℃干燥得多拉菌素9.6g,收率为96%,纯度为99.01%。
对所得多拉菌素晶体物进行XRD测试及HPLC检测,结果:
图1是实施例1得到的多拉菌素晶型X-射线粉末衍射图,图4为纯度检测HPLC图谱。从图4可以看出其主峰归一化纯度为99.16%。从图1可以看出,XRD衍射2θ值=6.5°、9.4°、10.5°、11.2°、11.5°、12.7°、13.4°、13.9°、14.4°、15.2°、15.7°、16.7°、17.1°、17.6°、18.2°、18.5°、19.1°、19.8°、21.3°、21.8°、22.3°、22.7°、25.3°、27.2°、30.4°处有专属特征吸收峰。
实施例2
取多拉菌素10g,加入丙酮30ml,升温至40℃溶解,减压浓缩至2倍体积,加入20ml 乙腈,继续浓缩至2倍体积,再加入10ml乙腈,继续浓缩至1-2倍体积,降温至-5-0℃,保温搅拌1小时,过滤,滤饼于50-60℃干燥得多拉菌素9.5g,收率为95%,纯度为99.54%。
对所得多拉菌素晶体物进行XRD测试及HPLC检测,结果:
图2是实施例2得到的多拉菌素晶型X-射线粉末衍射图,图5为纯度检测HPLC图谱。从图2中可以看出,在2θ值为4.4°、6.1°、6.3°、8.7°、9.3°、10.7°、11.0°、11.2°、11.5°、 12.5°、13.3°、13.8°、14.2°、14.4°、16.9°、17.6°、18.0°、18.2°、18.5°、18.9°、19.8°、21.4°、 22.0°、22.5°、26.8°、28.1°、33.0°、45.1°处有专属特征吸收峰。从图5可以看出其主峰归一化纯度为99.54%。
实施例3
取多拉菌素10g,加入甲醇50ml,升温至40℃溶解,减压浓缩至2倍体积,加入20ml 甲苯,继续浓缩至2倍体积,再加入10ml甲苯,继续浓缩至约1倍体积,降温至10-15℃,过滤,滤饼于60-80℃干燥得多拉菌素9.8g,收率为98%,纯度为99.45%。
对所得多拉菌素晶体物进行XRD测试,结果:
XRD衍射2θ值=6.4°、9.3°、10.3°、11.0°、12.4°、13.3°、14.2°、15.0°、15.6°、16.6°、 17.0°、17.3°、17.7°、18.2°、19.0°、22.3°、27.0°处有专属特征吸收峰。
实施例4
取多拉菌素10g,加入二氯甲烷30ml,升温至35℃溶解,减压浓缩至1倍体积,加入 15ml乙腈,继续浓缩至2倍体积,再加入20ml乙腈,继续浓缩至约1倍体积,降温至-5-0℃,过滤,滤饼于40-50℃干燥得多拉菌素9.9g,收率为99%,纯度为99.68%。
对所得多拉菌素晶体物进行XRD测试,结果:
XRD衍射2θ值=4.5°、6.2°、6.5、9.5°、10.4、11.2°、12.6°、13.4°、13.9、14.4°、16.9°、 17.7°、18.1°、18.4°、18.7°、20.0°、21.5°、22.7°、26.9°、28.2°、33.2°、45.3°处有专属特征吸收峰。
实施例5
取多拉菌素10g,加入乙醇35ml,升温至50℃溶解,减压浓缩至1倍体积,20ml正庚烷,继续浓缩至2倍体积,再加入10ml正庚烷,继续浓缩至约1倍体积,降温至0-5℃,过滤,滤饼于50-60℃干燥得多拉菌素9.9g,收率为99%,纯度为99.72%。
对所得多拉菌素晶体物进行XRD测试,结果:
XRD衍射2θ值=6.5°、9.4°、10.5°、11.2°、11.5°、12.6°、13.3°、13.8°、14.6°、15.2°、 15.9°、16.7°、17.1°、17.6°、18.2°、18.6°、19.2°、19.7°、21.4°、21.8°、22.4°、22.7°、25.5°、 27.3°、30.5°处有专属特征吸收峰。
实施例6稳定性研究
对该多晶型进行稳定性研究,结果如下:
表1多拉菌素晶型稳定性研究
实验结果表明:
在上表实验条件下,本发明所得的多拉菌素晶型,含量、外观及晶型等均无明显变化,稳定性好。且无吸湿性,可以更好的应用到药物制剂中。
本发明提出的多拉菌素晶型A、晶型B及其制备方法已通过实施例进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的多拉菌素多晶型及其制备方法进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明的精神、范围和内容中。
- 还没有人留言评论。精彩留言会获得点赞!