一种艾日布林中间体ER806060的合成方法与流程
一种艾日布林中间体er806060的合成方法
技术领域
[0001]
本发明属于化学合成技术领域,具体涉及一种艾日布林中间体er806060(cas:871348-08-2)的合成方法。
背景技术:
[0002]
艾日布林作为一种抗癌药,此化合物可以由三个片段通过一系列的化学反应组装而成,其关键的一个片段就是er806067(cas:871348-24-2)。此片段合成的一个前导化合物为er806060。
[0003]
目前已有的专利或者文献(wo2005/118565 a1)在合成er806060过程中:存在操作繁琐、收率低、三废较多、反应稳定性较差,产生不可控的杂质等缺点,以致生产风险性较大、成本高,不适用于工业生产。
[0004]
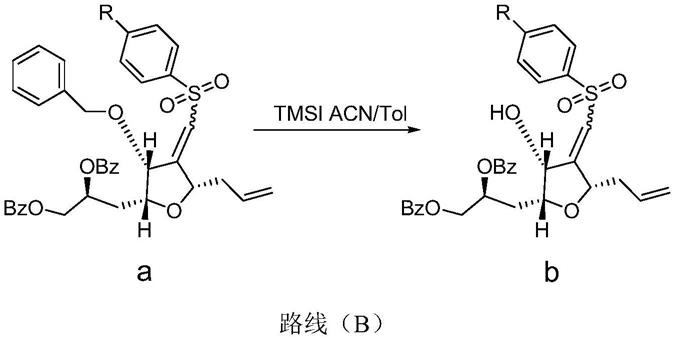
其中专利文献(wo2005/118565 a1)所记载的方法是以化合物a为原料,经过tmsi来脱去保护基苄基来合成化合物b,其合成过程如路线(b)所示:
[0005][0006]
其中,r为h或者ch3或者ch3ch2。
[0007]
该方法采用tmsi来脱掉保护基苄基有明显的、难以避免的缺陷:1.反应中所使用的tmsi容易不可控的产生难以除去的多种杂质;2.此反应是在高温下反应,反应完需立即将温,条件比较苛刻,难以实现工业化;3.此反应体系用氨水淬灭,淬灭搅拌的时间比较长(需要过夜),明显提高了工业化的生产时间,大幅提高车间成本。
技术实现要素:
[0008]
为了克服现有技术的上述缺陷,本发明首次提供了一种新型的关于艾日布林中间体er806060的工业化合成方法。本发明的合成方法以式a化合物作为原料,经过路易斯酸脱苄基保护反应合成艾日布林中间体er806060。本发明具有反应稳定性高,操作简捷,经济环保,产率高(约80%),不可控杂技少,适合工业化生产等优点。
[0009]
本发明提出了一种如式(b)所示的艾日布林中间体er806060的合成方法,其合成过程如路线(a)所示,
[0010][0011]
其中,r为h或者c1-c6的烷基;
[0012]
优选地,r为h或者ch3或者ch3ch2。
[0013]
本发明所述的艾日布林中间体er806060的合成方法,具体步骤如下:
[0014]
在有机溶剂中,式a化合物与路易斯酸试剂发生脱苄基保护基反应,得到式b化合物。
[0015]
此步骤中,所述有机溶剂为本领域常用非质子性溶剂,选自二氯甲烷、1,2-二氯乙烷、四氢呋喃(thf)、四氯化碳、氯苯、甲苯、二甲苯等中的一种或多种;优选地,为二氯甲烷。
[0016]
此步骤中,所述路易斯酸试剂为四氯化钛、三氯化铁、四氯化锡、三氯化铝三溴化硼等中的一种或者多种;优先地,为四氯化钛。
[0017]
此步骤中,所述式a化合物、路易斯酸试剂的摩尔比为1:(1~10);优选地,为1:(1~5);进一步优选地,为1:5。
[0018]
此步骤中,所述脱苄基保护基反应的温度为0~140℃;优选地,为30℃。
[0019]
此步骤中,所述脱苄基保护基反应的时间为1-10h;优选地,为3-8h;进一步优选地,为5h。
[0020]
此步骤中,所述反应稳定性高,操作简单,反应产率高(约80%),风险性小。
[0021]
本发明得到所述式b化合物前还包括纯化步骤,将脱苄基保护基反应后得到的式b化合物粗品,进行纯化步骤,得到所述式b化合物。
[0022]
其中,所述纯化的具体步骤为式b化合物粗品经过快速柱层析(硅胶(投料量的5wt),洗脱剂为石油醚/乙酸乙酯=4/1),收集含产品式b化合物的成份,浓缩即可以得到式b化合物。
[0023]
同时本发明也对此反应的可能反应机理进行了阐述,所述反应的反应机理如下式(c)所示:
[0024][0025]
式a化合物与四氯化钛先生成a1,并离去一个氯负离子,此氯负离子进攻苄基碳正离子生成中间体a2,并脱去苄氯得到中间体a3,淬灭得到式b化合物。
[0026]
本发明还提供了由上述方法制备得到的艾日布林中间体er806060。
[0027]
本发明提出了艾日布林中间体er806060,其结构如式(b)所示:
[0028][0029]
其中,r为h或者c1-c6的烷基;
[0030]
优选地,r为h或者ch3或者ch3ch2。
[0031]
本发明区别于现有技术或最主要的创新点为:
[0032]
本发明采用路易斯酸试剂如四氯化钛作为脱苄基保护基试剂的合成方法,与现有的合成方法相比有显著的优势:(1)本发明采用路易斯酸试剂如四氯化钛作为脱苄基保护基反应,能够避免诸如tmsi的使用所带来的操作繁琐、风险性大、不可控杂质的产生而导致的成本高等问题;(2)采用路易斯酸试剂如四氯化钛作为脱苄基保护基反应,能够避免因使用tmsi的后处理所需要比较长的淬灭时间,进而降低生产成本。本发明减少了反应中不可控杂质的产生、缩短了反应和处理时间、降低了操作难度,明显降低了成本。
[0033]
在一具体实施方式中,所述式(b)的艾日布林中间体er806060的工业化合成路线如下所示:
[0034][0035]
其中,r为h或者c1-c6的烷基;
[0036]
优选地,r为h或者ch3或者ch3ch2。
[0037]
本发明的有益效果在于,本发明反应时间短,处理时间短,操作简捷,废液可以循环套用,产率高(约80%),不可控杂质少,生产成本低,非常适用于工业化规模生产。
[0038]
说明书中用到简称对应全称对应表
[0039]
entry缩写全称1dcm二氯甲烷2tmsi三甲基碘硅烷3ticl4四氯化钛
具体实施方式
[0040]
结合以下具体实施例,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
[0041]
实施例1
[0042]
化合物b的合成(以r=h为例):
[0043]
方案1:
[0044]
氮气保护下,于100l反应釜中加入二氯甲烷(60l,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(6kg,9.19mol),30℃下滴加四氯化钛(8.72kg,45.95mol),30℃反应5小时,hplc或tlc检测,反应结束。降温到10℃,加入水(30l,5v),静置分液,水相用二氯甲烷(6l,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(6l,1v)洗涤,有机相减压浓缩到1.5v(9l),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物(4.14kg,7.35mol)。
[0045]1h nmr(400mhz,chloroform-d)δppm 2.15-2.35(m,5h)3.09-3.21(m,2h)3.40(dt,j=8.50,5.65hz,1h)3.98(q,j=6.44hz,1h)4.37-4.44(m,1h)4.54-4.64(m,2h)4.77-4.94(m,2h)5.54-5.79(m,2h)7.43(q,j=7.50hz,4h)7.51-7.66(m,4h)7.67-7.75(m,1h)7.93(d,j=7.34hz,2h)8.04(dd,j=13.45,7.34hz,4h)。
[0046]
此方案通过四氯化钛脱苄基保护基反应合成式b化合物,收率高(80%),而且操作流程简变,反应安全性高,不可控杂质无,并且此步骤的后处理过程中产生的废溶剂(石油醚以及乙酸乙酯)可以回收利用,产生的三废较少,相比于现有专利文献中记载的方法,本发明优势明显,适合于工业化生产。
[0047]
同时本发明也对此步进行了相关的优化,具体实施方案如下:
[0048]
方案1-1:
[0049]
氮气保护下,于500ml反应瓶中加入1,2-二氯乙烷(200ml,10v)或者甲苯(200ml,10v)或者氯苯(200ml,10v)或者四氯化碳(200ml,10v)或者二氯甲烷(200ml,10v)或者二甲苯(200ml,10v)或者四氢呋喃(200ml,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol),20℃下滴加四氯化钛(28.45g,0.15mol),30℃反应5小时,hplc或tlc检测,反应结束。降温到10℃,加入水(100ml,5v),静置分液,水相用二氯甲烷(20ml,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(20ml,1v)洗涤,有机相减压浓缩到1.5v(30ml),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物。
[0050]
二氯甲烷体系得到的式b化合物(13.79g,0.0245mol);甲苯体系得到的式b化合物(8.62g,0.015mol);氯苯体系得到的式b化合物(6.90g,0.012mol);二甲苯体系得到式b化合物(0.56g,0.001mol);1,2-二氯甲烷体系得到的式b化合物(4.50g,0.008mol);四氯化碳体系得到的式b化合物(2.81g,0.005mol);四氢呋喃体系无式b化合物的生成。
[0051]
因此在本方案中确定的反应溶剂优选为二氯甲烷。
[0052]
方案1-2:
[0053]
氮气保护下,于500ml反应瓶中加入二氯甲烷(200ml,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol),20℃下滴加四氯化钛(28.45g,0.15mol)或者加入四氯化锡(39.01g,0.15mol)或者三氯化铁(24.33g,0.15mol)或者三氯化铝(20.00g,0.15mol)或者三溴化硼(37.58g,0.15mol),30℃反应5小时,hplc或tlc检测,反应结束。降温到10℃,加入水(100ml,5v),静置分液,水相用二氯甲烷(20ml,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(20ml,1v)洗涤,有机相减压浓缩到1.5v(30ml),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物。
[0054]
四氯化钛(28.45g,0.15mol)体系得到的式b化合物(13.79g,0.0245mol);四氯化锡(39.01g,0.15mol)体系得到的式b化合物(11.02g,0.0196mol);三氯化铁(24.33g,0.15mol)体系得到的式b化合物(9.65g,0.0172mol);三氯化铝(20.00g,0.15mol)体系未得到的式b化合物;三溴化硼(37.58g,0.15mol)体系得到的式b化合物(9.00g,0.016mol)。
[0055]
因此在本方案中确定的路易斯酸优选为四氯化钛。
[0056]
方案1-3:
[0057]
氮气保护下,于500ml反应瓶中加入二氯甲烷(200ml,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol),20℃下滴加四氯化钛(39.83g,0.21mol)或者四氯化钛(28.45g,0.15mol)或者四氯化钛(17.44g,0.09mol),30℃反应5小时,hplc或tlc检测,反应结束。降温到10℃,加入水(100ml,5v),静置分液,水相用二氯甲烷(20ml,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(20ml,1v)洗涤,有机相减压浓缩到1.5v(30ml),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物。
[0058]
四氯化钛(17.44g,0.09mol)体系得到的式b化合物(10.34g,0.018mol);四氯化钛(28.45g,0.15mol)体系得到的式b化合物(13.79g,0.0245mol);四氯化钛(39.83g,0.21mol)体系得到的式b化合物(13.79g,0.0245mol)。
[0059]
因此在本方案中路易斯酸四氯化钛/化合物a的摩尔比暂定为5:1。
[0060]
方案1-4:
[0061]
氮气保护下,于500ml反应瓶中加入二氯甲烷(200ml,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol),20℃下滴加四氯化钛(28.45g,0.15mol),30℃反应3小时或者反应5小时或者反应7小时,hplc或tlc检测,反应结束。降温到10℃,加入水(100ml,5v),静置分液,水相用二氯甲烷(20ml,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(20ml,1v)洗涤,有机相减压浓缩到1.5v(30ml),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物。
[0062]
反应3小时还有原料剩余,体系得到的式b化合物(8.62g,0.015mol);反应5小时得到的式b化合物(13.79g,0.0245mol);反应7小时得到的式b化合物(13.79g,0.0245mol)。
[0063]
因此在本方案中最佳反应时间暂定为5小时。
[0064]
方案1-5:
[0065]
氮气保护下,于500ml反应瓶中加入二氯甲烷(200ml,10v),再加入(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tetr ahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol),20℃下滴加四氯化钛(28.45g,0.15mol),20℃反应5小时或者30℃反应5小时或者40℃反应5小时,hplc或tlc检测,反应结束。降温到10℃,加入水(100ml,5v),静置分液,水相用二氯甲烷(20ml,1v)萃取两次,合并有机相,饱和碳酸氢钠水溶液(20ml,1v)洗涤,有机相减压浓缩到1.5v(30ml),快速柱层析,洗脱剂为石油醚/乙酸乙酯,得到式b化合物。
[0066]
20℃反应5小时还有原料剩余,体系得到的式b化合物(5.63g,0.01mol);30℃反应5小时得到的式b化合物(13.79g,0.0245mol);40℃反应5小时得到的式b化合物(13.79g,0.0245mol)。
[0067]
因此在本方案中最佳反应温度暂定为30℃。
[0068]
方案2(wo2005/118565a1):
[0069]
此专利文献提到的方法是以化合物a为原料,使用tmsi作为脱苄基保护基合成化合物b,其合成过程如路线(b)所示:
[0070][0071]
具体实施例如下:
[0072]
化合物b的合成:
[0073]
氮气保护下,化合物a(s)-3-((2r,3r,5s))-5-烯丙基-3-(苄氧基)-4-((苯基磺酰基)亚甲基)四氢呋喃-2-基)丙烷-1,2-二基二苯甲酸酯((s)-3-((2r,3r,5s)-5-allyl-3-(benzyloxy)-4-((phenylsulfonyl)methylene)tet rahydrofuran-2-yl)propane-1,2-diyl dibenzoate)(20g,0.03mol)溶于甲苯(50ml,2.5v),乙腈(50ml,2.5v),30℃下滴加tmsi(24g,0.12mol),加毕,体系升温到60℃,在60℃反应2小时,tlc或者hplc反应完,将体系降温到-15℃,然后滴加25%的氨水(100ml,5v),30℃下搅拌过夜,分液,水相用甲苯(100ml,5v)萃取,合并有机相,有机相再依次用10%的亚硫酸钠水溶液(100ml,5v)洗涤,1n的盐酸(100ml,5v)洗涤,5%碳酸氢钠(100ml,5v)洗涤,饱和食盐水(100ml,5v)洗涤,无水硫酸钠干燥(4g,0.2wt),55℃浓缩干得粗品,柱层析得化合物b(12.07g,0.021mol),收率为
70%。
[0074]1h nmr(400mhz,chloroform-d)δppm 2.15-2.35(m,5h)3.09-3.21(m,2h)3.40(dt,j=8.50,5.65hz,1h)3.98(q,j=6.44hz,1h)4.37-4.44(m,1h)4.54-4.64(m,2h)4.77-4.94(m,2h)5.54-5.79(m,2h)7.43(q,j=7.50hz,4h)7.51-7.66(m,4h)7.67-7.75(m,1h)7.93(d,j=7.34hz,2h)8.04(dd,j=13.45,7.34hz,4h)。
[0075]
此步的缺点就是使用tmsi脱苄基保护基时,反应必须在高温下进行,一旦反应完必须立即降温,或者在原料没有反应完就必须提前降温淬灭,而且容易产生不可控的杂质碘化化合物,而且此步操作产生的废水量也比较大,从而进一步增加了生产成本,同时也增加了操作的繁琐性。
[0076]
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1