一类含多“锚点”的标记化合物及其制备方法与应用与流程
1.本发明属于生物标记及生物医药领域,具体涉及一类含多“锚点”的高效生物大分子标记化合物及其制备方法与应用。
背景技术:
2.亚甲基苯醌(qms)是一类反应性很高的活性中间体,其在自然界中应用最为广泛的结构为邻位亚甲基苯醌(o-qms)和对位亚甲基苯醌(p-qms)。例如,许多动物和植物利用qms作一种防御手段;qms参与阿霉素及其衍生物表阿霉素的抗癌过程。此外,qms也因其具有极高的亲电性,极易受到来自生命系统中亲核试剂的进攻,从而具有非常强烈的捕捉生物大分子并与之建立共价键的潜力。介于此性质,qms也被广泛应用于生命科学领域的研究中,用于开发高效、稳定的生物偶联试剂。例如,早在1990年有科学家将o-qms活性中间体“捕捉”并“锚定”生命大分子中亲核基团的化学过程引入葡萄糖苷酶活性抑制剂的工作机制中。随后,基于该类策略的抑制剂、捕获剂及其检测手段也被进一步应用于酶、抗体及特异性细胞分选的研发中。由于参与生命过程及构成生物体的物质基础即复杂又繁多,致使这种策略在很大程度上受到qms活性中间体低特异性的限制。qms作为“自锚定”式成像试剂也已广泛应用于磷酸酶、糖苷酶、脂肪酶、硫酸酯酶、β-内酰胺酶等多种重要生物酶的活性检测。然而目前基于qm 的探针分子对生物大分子的标记效率较低。而且现有研究仍然未能给出一个令人满意的答案。
3.综上所述,本领域迫切需要开发一种具有高效率、高灵敏度的生物大分子共价标记试剂。
技术实现要素:
4.本发明的目的就是提供一种具有高效率、高灵敏度的生物大分子共价标记试剂。
5.在本发明的第一方面,提供了一种式i化合物,
[0006][0007]
式i中,
[0008]
a选自下组:靶向识别基团(较佳地,为磷酸、糖苷、脱氧核苷、肽类化合物)、离去基团(较佳地,选自:羟基保护基团、氨基保护基团、巯基保护基团)、或二次或多次释放功能结构;
[0009]
a基团中,所述靶向识别基团为能够被生物大分子识别并与靶向识别基团相互作
用,从而产生酚羟基、酚巯基或氨基的基团;所述离去基团是指在对光、热、或化学诱导敏感且能够使a-x间共价键断裂从而产生酚羟基、酚巯基或氨基的基团;所述二次或多次释放功能的结构是指能够经触发剂激活后水解并释放出o,p-qm中间体的基团;且所述二次或多次释放功能的结构如式a所示
[0010][0011]
式a中,a’选自下组:靶向识别基团、离去基团;
[0012]
x为o、nh或s;
[0013]
lg是能够与偕二氟(chf2)、芳环母核共同构成多“锚点”官能团;
[0014]
l为连接基团;
[0015]
r选自下组:报告基团、治疗基团;其中,所述报告基团是指能够被探测荧光、放射和/或磁信号的基团,所述治疗基团是指具有治疗功能的药物分子及药物前体;
[0016]
在另一优选例中,
[0017]
a选自下组:磷酸基(-h2po3);糖苷;脱氧核苷;羟基、氨基或巯基保护基团(较佳地,光敏性保护基团;更佳地,且r”选自下组: c1-c6烷基、c1-c6卤代烷基);
[0018]
x选自下组:o、nh、s;
[0019]
lg选自下组:f、cl、br、oac、oconhr
b
、r
b
so2、-nr
b3+
;且r
b
各自独立地选自下组:c1-c6烷基、取代或未取代的苯基(较佳地,c6h5no2);
[0020]
l为连接基团;
[0021]
r选自下组:报告基团、药物基团;其中,所述报告基团是指能够被探测荧光、放射和/或磁信号的基团,所述药物基团是指具有治疗功能的药物分子及药物前体;
[0022]
除非特别说明,所述的取代是指基团中一个或多个氢被选自下组的取代基所取代:卤素(较佳地,f、cl、br、i)、c1-c6烷基、c1-c6卤代烷基。
[0023]
在另一优选例中,l为-wa-l
1-wb-;且wb与r连接;l基团中,
[0024]
wa和wb各自独立地选自下组:无、o、s、nr
a
、co、coo、so、so2、 co-nr
a
、nr
a-co、so-n(r
a
)、n(r
a
)-so、nr
a-coo、coo-nr
a
、nr
a-so2、so
2-nr
a
、 cs-nr
a
、nr
a-cs、n(r
a
)-co-nr
a
、-(ch2)
0、1或2-含1-3个氮杂原子的五或六元环-(ch2) 0、1或2-(较佳地,);
[0025]
l1为由选自下组的一个或多个单元结构组成的连接基团:取代或未取代的 c1-c4亚烷基、含1-3个氮杂原子的五或六元环(较佳地,为)、(取代或未取代的c1-c2亚烷基)-o、(取代或未取代的c1-c2亚烷基)-o-(取代或未取代的c1-c2 亚烷基)、(取代或未取代的c1-c2亚烷基)-s、(取代或未取代的c1-c2亚烷基)-s-(取代或未取代的c1-c2亚烷基)、nr
a
、co、coo、co-nr
a
、nr
a-co;
[0026]
r
a
各自独立地选自下组:h、c1-c6烷基、c1-c6卤代烷基,或取代或未取代的 c3-c6环烷基(较佳地,r
a
为h)。
[0027]
在另一优选例中,所述药物基团是指衍生自选自下组的药物的:抗生素(较佳地,头孢、万古霉素);阿维巴坦;舒尼替尼,金刚烷胺、阿昔洛韦。
[0028]
在另一优选例中,能够被探测放射基团是指被
18
f标记的基团。
[0029]
在另一优选例中,a选自下组:磷酸基(-h2po3)、和且r”选自下组:c1-c6烷基、c1-c6卤代烷基;较佳地,a为磷酸基。
[0030]
在另一优选例中,x为o。
[0031]
在另一优选例中,lg选自下组:f、oconhr
b
;且r
b
各自独立地选自下组: c1-c6烷基(较佳地,et、me)、取代或未取代的苯基(较佳地,c6h5no2)。
[0032]
在另一优选例中,wa为-(ch2)
0、1或2-含1-3个氮杂原子的五或六元环-(ch2)
0、1或2-(较佳地,)。
[0033]
在另一优选例中,wb选自下组:无、nr
a-co。
[0034]
在另一优选例中,l1选自下组:-(ch2)
n1-nhco-(ch
2-o-ch2)
n2-(ch2)
n1-、
ꢀ-
(ch2)
n3-、-(ch2)
n1-ar
1-(ch2)
n1-nhco-(ch
2-o-ch2)
n2-(ch2)
n1-。
[0035]
在另一优选例中,r为报告基团;较佳地,r为能够发出荧光的基团。
[0036]
在另一优选例中,所述能够发出荧光的基团为荧光素或荧光增强型染料。
[0037]
在另一优选例中,r选自下组:
[0038][0039]
在另一优选例中,l选自下组:
[0040]-(ch2)
n1-ar
1-(ch2)
n1-nhco-(ch
2-o-ch2)
n2-(ch2)
n1-nhco-、
[0041]-(ch2)
n1-ar
1-(ch2)
n3-、
[0042]-conh-(ch2)
n1-ar
1-(ch2)
n1-nhco-(ch
2-o-ch2)
n2-(ch2)
n1-nhco-;
[0043]
其中,n1=0、1、2或3;n2=1、2、3、4或5;n3=2、3、4、5、6、7、8或9;
[0044]
ar1为含1-3个氮杂原子的五或六元环(较佳地,)。
[0045]
在另一优选例中,l选自下组:
[0046][0047]
在另一优选例中,所述化合物如式iv所示,
[0048][0049]
其中,lg、l、r如前定义。
[0050]
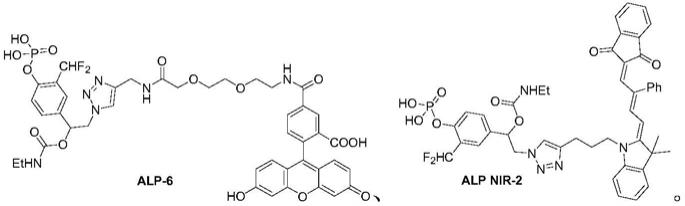
在另一优选例中,a、x、lg、l和r的为表1中具体化合物(较佳地,alp-6 和alp nir-2)中对应的基团。
[0051]
在另一优选例中,式i化合物选自下组:
[0052][0053]
在本发明的第二方面提供了如第一方面所述化合物的制备方法,包括步骤:
[0054]
(i)使式ia化合物与n
2-l
2-r(ib)反应,从而得到式i化合物;
[0055][0056]
l2为≡-l
1-wb(≡是指碳碳三键),
[0057]
a、x、lg、l1和wb如前定义。
[0058]
在另一优选例中,式i化合物如式iv所示,且制备式iv化合物的方法包括步骤:
[0059][0060]
(i)使式iva化合物与n
2-l
2-r(ib)反应,从而得到式iv化合物。
[0061]
在另一优选例中,式ia化合物或式iva化合物为化合物4
[0062][0063]
在另一优选例中,提供了一种化合物4的制备方法,包括步骤
[0064][0065]
(1)制备化合物1:
[0066]
(1a)在惰性溶剂1a(较佳地,dmf)中,使化合物1a(2-溴-1-[4-羟基-3-(羟甲基)苯基]-乙-1-酮)与nan3反应,得到叠氮化中间体;和
[0067]
(1b)在惰性溶剂1b(较佳地,超干(无水)乙腈)中,在n,n-二异丙基乙胺 (dipea)和4-二甲氨基吡啶(dmap)的存在下,使步骤(1a)得到的叠氮化中间体与亚磷酸二乙酯(5)和四氯化碳反应,得到化合物1;
[0068]
(2)制备化合物2:
[0069]
(2a)在惰性溶剂2a(较佳地,二氯甲烷)中,使化合物1与戴斯-马丁氧化剂 (dmp)反应,得到化合物2的中间体,
[0070]
(2b)在惰性溶剂2b(较佳地,乙醇)中,使步骤(2a)得到的中间体与二乙胺基三氟化硫(dast)反应,得到化合物2;
[0071]
(3)制备化合物3:
[0072]
(3a)在惰性溶剂3a(较佳地,甲醇)中,使化合物2与nabh4,从而得到化合物3的中间体;
[0073]
(3b)在惰性溶剂3b(较佳地,三乙胺(tea))中,使步骤(3a)的中间体与异氰酸乙酯反应,从而得到化合物4;
[0074]
(4)制备化合物4:
[0075]
在惰性溶剂4(较佳地,无水乙腈)中,使化合物4与三甲基溴硅烷(tmsbr) 反应,得到化合物4。
[0076]
在本发明的第三方面,提供了一种检测试剂或检测组合物,所述的检测试剂或检测组合物包括如第一方面所述的化合物和检测学上可接受的载体。
[0077]
在本发明的第四方面,提供了一种药物组合物,所述药物组合物包括如第一方面所述的化合物和检测学上可接受的载体。
[0078]
在本发明的第五方面,提供了一种中间体,所述的中间体如式ia所示,
[0079][0080]
式ii中,a、x、lg如前定义。
[0081]
在另一优选例中,所述中间体如式iva所示
[0082][0083]
在本发明的第六方面提供了一种体外非治疗性抑制alp过表达的方法,包括步骤:使对象与式i化合物接触,从而抑制alp过表达。
[0084]
在另一优选例中,所述的对象为细胞。
[0085]
在本发明的第七方面,提供了一种治疗和/或预防与alp过表达相关的疾病的方法,包括步骤:向需要的对象施用如第一方面所述的化合物或如第四方面所述的药物组合物。
[0086]
在另一优选例中,所述与alp过表达相关的疾病为肿瘤。
[0087]
在本发明的第八方面,提供了一种标记过表达alp细胞的方法,其特征在于,包括步骤:使对象与式i化合物接触,从而抑制alp过表达。
[0088]
在本发明的第九方面,提供了一种诊断患者体内与alp过表达相关的疾病的方法,包括步骤:向需要的对象施用如第一方面所述的化合物或如第三方面所述的检测试剂或检测组合物。
[0089]
在另一优选例中,所述与alp过表达相关的疾病为肿瘤。
[0090]
在本发明的第十方面,一种如第一方面化合物的用途,其特征在于,(i) 用于对过表达alp的细胞进行标记、追踪和/或成像;和/或(ii)用于制备对过表达alp的细胞进行标记、追踪和/或成像的组合物;和/或(iii)用于制备用于治疗与alp过表达相关的疾病的药物或组合物。
[0091]
在另一优选例中,当式i化合物的用途为(i)和/或(ii)时,式i中r为报告基团。
[0092]
在另一优选例中,当式i化合物的用途为(iii)时,式i中r为治疗基团。
[0093]
在另一优选例中,所述的对过表达alp的细胞进行标记、追踪和/或成像是体外非治疗性的。
[0094]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例) 中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
[0095]
图1为荧光探针alp-6及其结构类似物在碱性磷酸酶(alp)激活下对蛋白质的标记图;
[0096]
图2为荧光探针alp-6及其结构类似物与牛血清蛋白(bsa)的非特异性结合图;
[0097]
图3为荧光探针alp-0、2、5和6的细胞成像;
[0098]
图4为荧光探针alp nir-2及其类似物在碱性磷酸酶(alp)激活下对蛋白质的标记及其与非酶蛋白质的非特异性结合图;
[0099]
图5为荧光探针alp nir-0、1和2的细胞成像图。图6显示了如第一方面所述的化合物或试剂的大致作用原理。
具体实施方式
[0100]
发明人经过广泛而深入地研究,意外地发现了一类具有多“锚点”特征的基于qm化学反应过程的探针分子或化合物或含该探针分子或化合物的标记试剂)。本发明的标记试剂能够多次产生qm活性中间体,因此能够多次标记蛋白并且能够减少因其他亲核试剂淬灭qm活性中间体而造成的低效标记,最终达到“高效、稳定”标记生物大分子的作用。基于此发明人完成了本发明。
[0101]
术语
[0102]
除非另有表述,术语“烷基”本身或作为另一取代基的一部分是指具有指定碳原子数的直链或支链烃基(即,c
1-8
表示1-8个碳)。烷基的例子包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、正戊基、正己基、正庚基、正辛基等。
[0103]
在本文中,除特别说明之处,术语“取代”指基团上的一个或多个氢原子被选自下组的取代基取代:卤素、未取代或卤代的c1-c6烷基。
[0104]
除非特别说明,本发明中,所有出现的化合物均意在包括所有可能的光学异构体,如单一手性的化合物,或各种不同手性化合物的混合物(即外消旋体)。本发明的所有化合物之中,各手性碳原子可以任选地为r构型或s构型,或r 构型和s构型的混合物。
[0105]
如本文所用,“共价标记化合物”、“标记化合物”、“共价标记分子”、“标记分子”、“式i化合物”、“本发明的探针分子”和“本发明的化合物”可以互换使用是指如第一方面所述的式i化合物。
[0106]
如本文所用,“衍生自”某一化合物或药物的基团(如本申请所述的药物基团)是指,药物或化合物上失去部分基团或原子(如1个h原子)从而形成能够与其他部分如本申请的l基团连接并保持或基本保持原始化合物或药物活性的基团。
[0107]
如本文所用,“二次或多次释放功能的结构”是指该结构中的触发试剂作用位点在触发试剂作用下,发生共价键的断裂(x-a断裂),产生第一个qm中间体,该中间体通过电子
重排快速离去,进而产生本专利所涉及的具有“多锚点”结构的核心qm中间体。一种示例性的结构如下式所示(其中,a’的定义同式i中的a基团,更佳地,a’选自:靶向识别基团、离去基团)。
[0108][0109]
亚甲基苯醌(qms)
[0110]
亚甲基苯醌(qms)是一类反应性很高的活性中间体,其在自然界中应用最为广泛的结构为邻位亚甲基苯醌(o-qms)和对位亚甲基苯醌(p-qms)。例如,许多动物和植物利用qms作一种防御手段;qms参与阿霉素及其衍生物表阿霉素的抗癌过程。此外,qms也因其具有极高的亲电性,极易收到来自生命系统中亲核试剂的进攻,从而具有非常强烈的捕捉生物大分子并与之建立共价键的潜力。介于此性质,qms也被广泛应用于生命科学领域的研究中,用于开发高效、稳定的生物偶联试剂。随后,基于该类策略的抑制剂、捕获剂及其检测手段也被进一步应用于酶、抗体及特异性细胞分选的研发中。由于参与生命过程及构成生物体的物质基础即复杂又繁多,致使这种策略在很大程度上受到qms活性中间体低特异性的限制。具体分析,可能是因为分子热运动导致具有高反应活性的qms中间体在靶蛋白催化位点形成后,迅速向周围扩散,若其与靶蛋白催化反应位点亲核基团迅速反应并连接;否则活化的qms中间体会与附近蛋白中的亲核部位发生共价连接。由此推想,前一过程可能会影响靶酶的结构与活性,继而产生靶酶活性抑制效果,可以用于蛋白的捕捉与酶抑制剂的研发;而后者则可降低了化学修饰给目标蛋白酶结构与活性带来的影响,不仅能更准确地反映酶的活性,而且能通过酶的“一点式触发”带来后期众多蛋白被标记的“瀑布式放大”效果,继而体现出qms中间体在化学修饰类成像试剂在生物应用中的优势。qms作为“自锚定”式成像试剂也已广泛应用于磷酸酶、糖苷酶、脂肪酶、硫酸酯酶、β-内酰胺酶等多种重要生物酶的活性检测及生物标记过程。
[0111][0112]
通常,基于qm的标记型探针分子在结构上包括:酶识别部分、潜在捕获基团、连接臂和报告基团。其中酶识别部位发挥寻找目标物质和启动“自锚定”过程的作用;潜在的捕获基团,即qm前体,发挥潜在生物分子“锚定”功能的,在蛋白质有效共价标记的过程中起着至关重要的作用。目前,传统的qms前体主要有两类:(1)o-单氟甲基苯酚或o-二氟甲基苯酚衍生物(o-qm前体);(2)4
-ꢀ
烷基酚,其苄位(4-烷基)连接离去基团(如氟、氨基甲酸酯和羧酸酯)(p-qm 前体)。两者均是在靶酶水解激活后,通过电子转移诱发离去基团的自发离开,原位释放高反应性qms活性中间体,继而引发探针分子与生物大分子间的共价连接。其中p-qm前体的潜在“锚点”远离酶的识别基团,预示着对探针分子结构的恰当修饰,不仅不会影响靶酶对探针的特异性识别,甚至可以探针增加分子的功能性。例如,将p-qm型荧光探针的
离取基团改造为荧光猝灭基团,从而得到荧光“off-on”的低背景、共价标记荧光探针。
[0113]
通过上面的阐述,亚甲基苯醌(qm)参与的基于酶活性的“自锚定”型生物大分子标记过程应当具有值得期待的应用与发展前景。然而,影响该策略实际应用的主要原因是分子对生物大分子的标记效率。其标记效率主要受限于qms的类型、形成的速率、亲电反应活性的高低,以及最终形成的共价偶联物的稳定性。如果标记效率相对较低,则明显阻碍该战略的实际应用,而现有研究仍然未能给出一个令人满意的答案。所以,研究qms结构与性能之间的联系,寻找高标记效率的“自锚定”型生物大分子标记试剂,可以有效改善试剂分子在实际应用中的信噪比,提高试剂的检测灵敏度,为研究生物标记技术和生物医药领域的拓展道路。
[0114]
多“锚点”[0115]
本发明中,如第一方面所述的化合物(式i)或试剂,该试剂(或化合物) 是通过连接臂将酶识别基团与具有荧光响应能力的报告基团相连得到的荧光 探针。该试剂(或化合物)在靶向识别基团作用下促使a-x键共价键的断裂, 并迅速生成具有高反应活性的醌亚甲基(qm)活性中间体,该活性中间体极易受 到生物大分子结构中亲核试剂的进攻,进而与生物大分子之间形成共价键。此 外,由于所述试剂结构总含有多个qm前体,使其具有多次激活并标记生物大 分子的能力,即多“锚点”特性,从而使所述试剂具有靶向性活化和高效地共价 标记生物大分子的性质,其作用原理大致如图6所示。
[0116][0117]
alp
[0118]
alp作为生物体内中重要的水解酶,不仅参与多种生物分子,包括核酸和蛋白质,以及其他小分子的脱磷酸化过程中;而且与许多疾病(如成骨细胞性骨癌、前列腺癌、肝炎)的发生、发展有着密切的关系,因此alp也被作为分子生物学甚至临床诊断中的重要生物标志物。故而对于alp活性和分布的检测方法也有不少研究,其中就不乏基于qm的“自锚定”荧光探针的开发。鉴于上述原因,本发明采用alp作为模型酶,设计荧光探针分子,深入研究了作为捕获单元的 qm活性中间体的结构与其标记效率间的关系,从中筛选并确定了具有多个离去基的qm前体结构作为捕获单元对于该类共价键标记试剂的实际应用较为有利。并且实现了对体内外alp的活性检测、alp过表达肿瘤细胞的活细胞成像。
[0119]
含多“锚点”的高效生物大分子共价标记化合物
[0120]
本发明提供了一种含多“锚点”的高效生物大分子共价标记化合物(或式i 化合物),所述试剂在碱性磷酸酶水解磷酸酯键的作用下生成高反应活性的o
-ꢀ
或p-qm中间体,使其与活化位点周围蛋白质结构中的亲核基团发生共价连接, 从而使所述试剂具有检测或标记alp的性质。
[0121]
典型地,本发明是提供了一类含多“锚点”的高效生物大分子共价标记化合物(即式i化合物),所述试剂如式i所示,
[0122]
[0123]
式中,
[0124]
a为能够被生物大分子(如酶、蛋白质及核酸等)识别并与之相互作用产生酚羟基(巯基或氨基)的靶向识别基团(如磷酸、糖苷、3-氧代正丁基或脱氧核苷),或对外加手段(如光、热或化学诱导)敏感并促使a-x间共价键断裂产生酚羟基(巯基或氨基)的基团(如羟基(oh)或羟基、巯基或氨基保护基团(如光敏性保护基团(光敏试剂)));亦或是具有“二次释放功能的结构”,例如,经催化水解首先形成一个或多个可离去的官能团(如,o,p-qm潜体),再通过该基团的离去从而释放出本文所述的o,p-qm中间体;
[0125]
x为o、nh或s;
[0126]
lg与偕二氟(chf2)、芳环母核共同构成多“锚点”官能团,其中lg可以是f、 cl、br、oac、oconhr
a
(r
a
=et、me、c6h5no2等)、砜(r
b
so2)或季铵盐(如
ꢀ-
nr
c3+
)等常见离去基团;
[0127]
l为以各种方式构建的连接基团,如酰胺键、酯键、醚键或以“点击化学”方式建立的连接基团等;
[0128]
r为具有荧光、放射或磁信号的报告基团,或为具有治疗功能的药物分子及药物前体。
[0129]
较佳地,所示光敏性保护基团为(uv光触发)或(nir 光触发)。
[0130]
较佳地,所述具有治疗功能的药物分子及药物前体是指衍生自,例如(但不限于)抗生素(头孢、万古霉素),用于抗菌、抵抗耐药;阿维巴坦,抑制丝氨酸类β-内酰胺酶抑制剂;舒尼替尼,选择性地靶向作用于多种受体酪氨酸激酶,阻断肿瘤生长并直接攻击肿瘤细胞,达到抗肿瘤目的;金刚烷胺、阿昔洛韦,用于抗病毒。
[0131]
在另一优选例中,多“锚点”的高效生物大分子共价标记化合物如式ii所示,其中,reporter为具有荧光、放射或磁信号的报告基团:
[0132][0133]
在另一优选例中,多“锚点”的生物大分子共价标记化合物如式iii所示,其中,drug为具有治疗功能的药物分子及药物前体:
[0134][0135]
进一步,所述试剂中的r为具有荧光的报告基团,报告基团为荧光素的荧光探针(alp-6)的结构为:
[0136][0137]
进一步,所述优先荧光探针能被碱性磷酸酶选择性识别并水解,并产生具有活泼化学性质的qm中间体,从而起到共价标记alp及其周围蛋白质的作用。
[0138]
进一步,alp-6能够特异性的被碱性磷酸酶(alp)水解激活,并生成o-或 p-qm中间体,随后该活性中间体与活化位点周围蛋白质结构中的亲核基团发生共价连接,从而起到标记及检测alp活性或标记alp过表达的肿瘤细胞的作用。
[0139]
进一步,所述试剂中的r为具有荧光的报告基团,报告基团为荧光增强型染料的荧光探针(alp nir-2)的结构为:
[0140][0141]
较佳地,alp nir-2在alp水解激活后,会通过与alp-6相同的历程,不仅能够共价标记于靶蛋白及其周围蛋白上,起到alp活性检测及alp过表达的肿瘤细胞的定位的作用;而且能够通过反应前后染料基团共轭体系建立,使探针的荧光强度较反应前有所增加,从
而达到相对提高目标信号强度、降低噪音、改善检测结果的目的。此外,由于所述荧光探针的发射波长位于近红外区域,所以可以利用近红外光组织透力强、组织损伤小的特点,为实现荷瘤小鼠瘤内 alp的活体成像打下基础。
[0142]
制备方法
[0143]
下面更具体地描述本发明式(i)结构化合物的制备方法,但这些具体方法不对本发明构成任何限制。本发明化合物还可以任选将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便地制得,这样的组合可由本发明所属领域的技术人员容易地进行。通常,在制备流程中,各反应通常在惰性溶剂中。反应时间通常为0.1小时-60小时,较佳地为0.5-48小时。
[0144]
典型地,本发明所提供的多“锚点”的高效生物大分子共价标记化合物(式i 化合物)的制备方法,包括步骤:
[0145][0146]
(i)在惰性溶剂i中,使式ia化合物(较佳地,式iva化合物如化合物4)与 n
2-l
2-r(ib)反应,从而得到式i化合物(或式iv化合物)。
[0147]
较佳地,所述惰性溶剂i选自下组:二甲基亚砜(dmso)、水,或其组合。
[0148]
较佳地,步骤(i)在维生素c、硫酸铜及三(3-羟丙基三唑甲基)胺的存在下使式ib化合物反应。
[0149]
较佳地,步骤(i)反应的反应时间为0.1~2小时(如0.5小时),和/或反应温度为室温(如20-30℃)。
[0150]
在一个具体实施例中,本发明的化合物4可参照下述流程制备:
[0151]
[0152]
在一个具体实施例中,本发明的式i化合物可通过下述流程制备:
[0153][0154]
该流程主要通过酚羟基的磷酸反应,引入酶识别位点;随后,通过对潜在的qm母核结构修饰,向qm母核结构引入了多个潜在离去基团;作为最终的共价键结合“锚点”;-接着完成了向潜在的qm母核上引入了可后期修饰的叠氮基团的目的,随后通过叠氮基与炔基间的“点击化学反应”实现酶识别基团、亲核试剂捕获基团与荧光报告基团间的连接。
[0155]
在一个具体实施例中,化合物1通过下述方法制备:
[0156]
将2-溴-1-[4-羟基-3-(羟甲基)苯基]-乙-1-酮与nan3加入反应瓶中,加入n,n
-ꢀ
二甲基甲酰胺(dmf)中;
[0157]
将反应体系在室温条件下与搅拌反应12小时后,重结晶得到原料的叠氮化中间体;
[0158]
将上述中间体与n,n-二异丙基乙胺(dipea)和4-二甲氨基吡啶(dmap)加入反应瓶中,加入超干乙腈,冷却至-20℃并滴加四氯化碳和亚磷酸二乙酯(5, diethyl phosphite);滴毕,将反应液转移至室温搅拌反应1小时,旋干溶剂后加入二氯甲烷溶解,饱和食盐水洗涤,无水硫酸镁干燥、浓缩后经硅胶柱层析快速分离纯化得到所述化合物1。
[0159]
在一个具体实施例中,化合物2通过下述方法制备:
[0160]
将化合物1和戴斯-马丁氧化剂(dmp)加入反应瓶中,加入二氯甲烷溶解;将反应体系在室温条件下反应2小时,加入饱和食盐水,分离有机相,水相用二氯甲烷萃取、合并有机相,无水硫酸镁干燥、浓缩,所得中间体粗产品未经进一步纯化,直接用于下一步实验;
[0161]
将上述中间体粗品加入反应瓶,加入重蒸二氯甲烷溶解,冰浴条件下滴加二乙胺基三氟化硫(diethylaminosulfur trifluoride,dast);
[0162]
滴毕,将反应体系与冰浴中搅拌反应2小时,随后用水淬灭,二氯甲烷萃取水层并收集有机相,有机相干燥、浓缩后经硅胶柱层析快速分离纯化得到所述化合物2;
[0163]
在另一个具体实施例中,化合物3通过下述方法制备:
[0164]
将化合物2加入反应瓶中,加入甲醇溶解并于冰浴下冷却,随后加入nabh4;将反应体系在冰浴条件下搅拌反应0.5小时,反应完毕加入饱和氯化铵水溶液终止反应,并用二氯甲烷萃取水相,合并的有机相经干燥、浓缩后得到反应中间体粗;
[0165]
将上述粗产品、异氰酸乙酯(6,ethyl isocyanate)与三乙胺(tea)加入反应瓶中,加入重蒸二氯甲烷溶解;
[0166]
将反应液加热回流12小时,反应结束后加水淬灭过量异氰酸乙酯,并用二氯甲烷萃取,合并有机相,无水硫酸镁干燥、浓缩,经硅胶柱层析得到所述化合物3。
[0167]
在另一个具体实施例中,化合物1通过下述方法制备:
[0168]
氮气保护下,将化合物3置于反应瓶中,加入超干乙腈溶解,冰浴条件下滴加三甲基溴硅烷(tmsbr);
[0169]
滴毕,将反应液置于室温下搅拌反应12小时,反应结束,用饱和氯化铵水溶液淬灭,随后反应液经反向c18柱分离纯化,冷冻干燥后获得所述化合物4。
[0170]
在另一个具体实施例中,式i化合物通过下述方法制备:
[0171]
在惰性溶剂中,使化合物4与化合物7(或8)反应,从而得到所述检测探针。
[0172]
优选地,在维生素c、硫酸铜及三(3-羟丙基三唑甲基)胺的存在下使化合物 4与化合物7(或8)反应。
[0173]
优选地,所述惰性溶剂为二甲基亚砜(dmso)和/或水。
[0174]
优选地,所述反应的反应时间为0.1~2小时(如0.5小时),和/或反应温度为室温(如20-30℃)
[0175]
优选地,所述方法还包括分离和/或纯化检测探针的步骤;较佳地,所述分离和/或纯化检测探针的步骤为用反相c18制备柱纯化,冷冻干燥。
[0176]
本发明的主要优点包括:
[0177]
(a)本发明的化合物具有多次产生qm活性中间体的能力,因此能多次标记蛋白;
[0178]
(b)本发明的化合物能够减少其他亲核试剂淬灭qm活性中间体而造成的无效标)。
[0179]
(c)本发明的化合物或标记试剂标记效率高、标记稳定。
[0180]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0181]
除非特别说明,在本发明的实施例中,1h-nmr、
13
c-nmr用bruker 400mz 型仪器测定,测定溶剂为氘代氯仿(cdcl3)、氘代二甲基亚砜(d
6-dmso),内标为四甲基硅烷(tms);所有溶剂均为色谱纯、分析纯或化学纯。
[0182]
实施例1.1化合物1的制备
[0183]
[0184]
将2-溴-1-[4-羟基-3-(羟甲基)苯基]-乙-1-酮(4g,16.3218mmol)与nan3(1.53 g,23.6mmol)加入反应瓶中,加入n,n-二甲基甲酰胺(dmf)中;
[0185]
将反应体系在室温条件下与搅拌反应12小时后,重结晶得到2.1168g原料的叠氮化中间体;
[0186]
将上述中间体(500mg,2.41mmol)与n,n-二异丙基乙胺(dipea,1.0495 ml,6.025mmol)和4-二甲氨基吡啶(dmap,29.9mg,0.241mmol)加入反应瓶中,加入超干乙腈,冷却至-20℃并滴加四氯化碳(2.32ml,24.1mmol)和亚磷酸二乙酯(5,diethyl phosphite,357μl,2.77mmol);
[0187]
滴毕,将反应液转移至室温搅拌反应1小时,旋干溶剂后加入二氯甲烷溶解,饱和食盐水洗涤,无水硫酸镁干燥、浓缩后经硅胶柱层析快速分离纯化得到化合物1(521mg,63%)。
[0188]
制得的化合物1的谱图特征为:1h nmr(400mhz,cdcl3)8.02(s,1h), 7.86(dd,j=8.5,2.1hz,1h),7.35(d,j=8.5hz,1h),4.72(s,2h),4.56(s,2h), 4.31
–
4.21(m,4h),1.38(t,j=7.1hz,6h).
13
c nmr(100mhz,cdcl3) 192.27,152.26(d,j=7.0hz),133.80(d,j=6.2hz),131.60,129.55,128.78, 120.62,65.44(d,j=6.2hz),59.28,54.88,16.07(d,j=6.6hz).
[0189]
实施例1.2化合物2的制备
[0190][0191]
将化合物1(190.37mg,0.55mmol)和戴斯-马丁氧化剂(dmp,251.2mg,0.592mmol)加入反应瓶中,加入二氯甲烷(2ml)溶解;
[0192]
将反应体系在室温条件下反应2小时,加入5ml饱和食盐水,分离有机相,水相用二氯甲烷萃取(5ml
×
3)、合并有机相,无水硫酸镁干燥、浓缩,所得中间体粗产品未经进一步纯化,直接用于下一步实验;
[0193]
将上述中间体粗品(180.5mg)加入反应瓶,加入5ml重蒸二氯甲烷溶解,冰浴条件下滴加二乙胺基三氟化硫(diethylaminosulfur trifluoride,dast,136 μl,1.109mmol);
[0194]
滴毕,将反应体系与冰浴中搅拌反应2小时,随后用2ml水淬灭,二氯甲烷萃取水层(5ml
×
3)并收集有机相,有机相干燥、浓缩后经硅胶柱层析快速分离纯化得到化合物2(144mg,72%).;
[0195]
制得的化合物2的谱图特征为:1h nmr(400mhz,cdcl3)8.17(s,1h), 8.05(dd,j=8.7,2.1hz,1h),7.64(d,j=8.8hz,1h),6.95(t,j=54.8hz,1h), 4.57(s,2h),4.32
–
4.19(m,4h),1.40
–
1.36(m,6h).
13
c nmr(100mhz,cdcl3)
[0196]
191.48,152.96(q,j=6.0hz),132.19,131.21,127.11(t,j=6.0hz),126.16(td, j=23.4,7.8hz),120.73(d,j=2.2hz),110.73(t,j=238.6hz),65.56(d,j=6.2 hz),54.96,16.14(d,j=6.6hz).
[0197]
实施例1.3化合物3的制备
[0198][0199]
将化合物2(201.6mg,0.555mmol)加入反应瓶中,加入5ml甲醇溶解并于冰浴下冷却,随后加入nabh4(31.5mg,0.833mmol);
[0200]
将反应体系在冰浴条件下搅拌反应0.5小时,反应完毕加入2ml饱和氯化铵水溶液终止反应,并用二氯甲烷(10ml
×
3)萃取水相,合并的有机相经干燥、浓缩后得到161.3mg反应中间体粗产品;
[0201]
将上述粗产品、异氰酸乙酯(6,ethyl isocyanate,175μl,2.208mmol)与三乙胺(tea,307μl,2.208mmol)加入反应瓶中,加入3ml重蒸二氯甲烷溶解;
[0202]
将反应液加热回流12小时,反应结束后加2ml水淬灭过量异氰酸乙酯,并用二氯甲烷(10ml
×
3)萃取,合并有机相,无水硫酸镁干燥、浓缩,经硅胶柱层析得到化合物3(115mg,47%)。
[0203]
制得的化合物3的谱图特征为:1h nmr(400mhz,cdcl3)7.60(s, 1h),7.50
–
7.42(m,2h),6.93(t,j=55.1hz,1h),5.88
–
5.83(m,1h),4.87(s, 1h),4.28
–
4.18(m,4h),3.61
–
3.43(m,2h),3.30
–
3.18(m,2h),1.36(t,j=7.1 hz,6h),1.15(t,j=7.2hz,3h).
13
c nmr(100mhz,cdcl3)154.82, 148.54(q,j=5.9hz),135.07,130.22,125.64(td,j=22.9,7.2hz),124.66(t,j= 5.8hz),120.41(d,j=1.8hz),111.06(t,j=237.7hz),73.86,65.11(d,j=6.2 hz),55.21,36.00,16.00(d,j=6.6hz),15.03.
[0204]
实施例1.4化合物4的制备
[0205][0206]
氮气保护下,将化合物3(115.2mg,0.264mmol)置于反应瓶中,加入超干乙腈溶解,冰浴条件下滴加三甲基溴硅烷(tmsbr,174μl,1.320mmol);
[0207]
滴毕,将反应液置于室温下搅拌反应12小时,反应结束,用饱和氯化铵水溶液淬灭,随后反应液经反向c18柱分离纯化,冷冻干燥后获得所述化合物4(34 mg,35%)。
[0208]
制得的化合物4谱图特征为:1h nmr(400mhz,d6-dmso)7.60(s, 1h),7.54(d,j=8.4hz,1h),7.48
–
7.39(m,2h),7.11(t,j=55.1hz,1h),5.80(t, j=5.6hz,1h),3.64(d,j=5.6hz,2h),3.00(qd,j=10.8,7.0hz,2h),1.00(t,j =7.2hz,3h).
13
c nmr(100mhz,d
6-dmso)154.93,149.29(q,j=6.0 hz),134.74,130.24,125.10(td,j=22.7,6.2hz),123.96
(t,j=4.7hz),120.97, 111.65(t,j=235.1hz),72.79,54.49,35.25,15.02.
[0209]
实施例1.5化合物7合成方法
[0210][0211]
将5-羧基荧光素(200.0mg,0.532mmol)、n-羟基琥珀酰亚胺(91.8mg,0.798 mmol)及edci(153.0mg,0.798mmol)置于反应瓶中,抽真空后氮气置换3次,在氮气保护下加入2ml干燥的dmf至完全溶解,于冰浴条件下搅拌反应4h,hplc监测反应完毕,向反应液中加入大量乙醚沉淀、离心,最终得到144.5mg粗产品7-1
[0212][0213]
将原料7-2(2.1631g,4.865mmol)与edci(1.3989g,7.297mmol)溶于8ml dcm中,冰浴条件下加入炔丙胺(347μl,5.838mmol),反应液缓慢升温至室温,继续搅拌,tlc监测至原料2完全消失后加入10ml饱和食盐水,用dcm萃取(5ml
×ꢀ
3),合并有机相,无水硫酸镁干燥,浓缩后经硅胶柱层析快速分离得1.1144g粗产品3。冰浴条件下,将100.0mg中间体7-3(0.333mmol)溶于2ml dcm/tfa/tips(10:9:1) 混合溶液中,冰浴下搅拌反应30min后hplc监测反应完全,旋干溶剂后,抽真空除去tfa,得到中间体4粗产品。称取23.67mg中间体1(0.05mmol)溶于40μl dmf中,随后分别加入30μl中间体7-4(c
dmf
=2m,0.06mmol)和42μl的tea(0.3mmol),室温下震荡反应1h后hplc监测反应完全,加入大量乙醚沉淀、离心,最终获得21.4mg 化合物7
[0214]
化合物7的低分辨质谱数据:ms(esi)m/z计算值c
30
h
26
n2o9(m+h)
+
559.2,实测值559.2;(m+na)
+
581.2,实测值581.2.
[0215]
实施例1.6荧光探针(alp-6)的制备
[0216][0217]
将化合物4(1.54mg,2.76μmol)、化合物7(c
dmf
=636.8mm,5μl,1.79μmol) 置于反应瓶中,加入二甲基亚砜(dmso,15l)、水、维生素c(1.3mg, 7.16mol)、硫酸铜(30g,0.18mol)及三(3-羟丙基三唑甲基)胺 (thpta,80g,0.18mol);
[0218]
将反应体系在室温下反应0.5小时,反应结束,用反相c18制备柱纯化,冷冻干燥,得到浅黄色的化合物,即所述荧光探针(alp-6,1.0mg,40%)。
[0219]
alp-6的谱图特征为:hrms(esi)m/z计算值c
42
h
41
f2n6o
15
p(m-h)
- 937.2263,实测值937.2256.
[0220]
化合物8可参照现有技术的方法制备。
[0221]
实施例1.7荧光探针(alp nir-2)的制备
[0222][0223]
将化合物4(2.90mg,7.63μmol)、化合物8(2.22mg,4.59μmol)置于反应瓶中,加入二甲基亚砜(dmso)、水、维生素c、硫酸铜及三(3-羟丙基三唑甲基)胺;
[0224]
将反应体系在室温下反应0.5小时,反应结束,用反相c18制备柱纯化,冷冻干燥,得到蓝色的化合物,即所述荧光探针(alp nir-2,1.0mg,31%)。
[0225]
谱图特征为:hrms(esi)m/z计算值c
46
h
44
f2n5o8p(m-h)-862.2823,实测值862.2819.
[0226]
实施例1.8荧光探针
[0227]
列表中的其他化合物可参照实施例1.5和/或1.6的方法制备。
[0228]
表1.alp检测探针的化学结构
[0229][0230][0231]
其中,a代表通式a,b代表通式b。
[0232]
alp-1、3、4、5和alp nir-1为单“锚点”alp检测探针,alp-2、6和alp nir-2为本发明的多“锚点”的检测探针(b),alp-0和alp nir-1为无“锚点
”ꢀ
alp检测探针。
[0233]
实施例2.1alp-6及其类似物在碱性磷酸酶(alp)激活下对蛋白质的标记
[0234]
实验条件:分别将7.5μm荧光探针(alp-6及其结构衍生物alp-0~5)与12.5 u/ml靶标酶alp(12.5μg)在37℃水浴中避光孵育1小时;或者进一步模拟生理环境,在上述反应液中同时添加1.5μg bsa作为靶酶的周围蛋白,于相同实验条件下避光孵育1小时。随后在150v的恒压条件,对上述蛋白质样品进行sds-page 凝胶电泳处理,并对蛋白质凝胶进行cy 2通道下的蛋白质胶内荧光扫描(下)及考马斯蓝染色(上)(结果参见图1)。
[0235]
实验结果:在模拟生理条件下,荧光探针alp-6及其类似物在碱性磷酸酶的特异性识别与水解作用后,不仅可以被激活并实现对靶蛋白alp及其周围蛋白质 (如bsa)的标记;而且由图1可见,不同的荧光探针,虽然其结构只存在微小差别,但是其对体系内蛋白质的标记效果却有明显不同;并且其中alp-3,4,5 和6对蛋白质的标记效果相对突出,通过结构比较与分析,上述有荧光分子均具有潜在p-qm结构。
[0236]
实验结论:通过对荧光探针分子结构进行修饰,可以获得对蛋白质共价标记效能相对理想的,且均含有潜在p-qm结构的“自锚定”荧光探针分子alp-3,4,5 和6。说明该类潜在p-qm结构对捕获基团与蛋白质亲核基团间共价键的建立更为有利;且它的存在能够更为有效地提高基于qm中间体的“自锚定”型荧光探针对生物大分子的标记效能。
[0237]
测试例2.2alp-6及其类似物与牛血清蛋白(bsa)的非特异性结合
[0238]
实验条件:分别将7.5μm荧光探针(alp-6及其结构衍生物alp-0~5)在无碱性磷酸酶(alp)催化条件下,仅与3μg bsa在37℃水浴中避光孵育1小时。随后在150v的恒压条件,对上述蛋白质样品进行sds-page凝胶电泳处理,并对蛋白质凝胶进行cy 2通道下的蛋白质胶内荧光扫描(下)及考马斯蓝染色(上)(结果参见图2)。
[0239]
实验结果:通过适当调节对比度,荧光探针alp-6及其类似物在无碱性磷酸酶水解激活的情况下,对体系中存在的非酶蛋白质bsa有不同程度的非特异性结合。其中探针alp-1、2、3及4的非特异性结合信号相对明显,而荧光探针alp-5、 6与bsa之间的非特异性结合非常微弱。
[0240]
实验结论:荧光探针与蛋白质分子间的这种非特异性结合比较常见,该类结合不仅是荧光探针对目标物质检测时出现假阳性的主要原因,也会大大降低荧光探针的真实性和实用性。图2结果说明荧光探针alp-5、6在检测靶酶alp时显示出来的荧光信号更为准确、真实,且在其应用中能够较为真实的反应该类荧光探针分子在酶激活后对蛋白质的共价标记效能。结合图1,荧光探针alp-5、6在酶活化后对蛋白质共价标记中的优秀表现,本发明一类具有良好信噪比、能够被酶特异性激活的高效“自锚定”型生物大分子共价标记化合物。
[0241]
实施例2.3使用alp-0、2、5和6进行细胞成像
[0242]
实验条件:在37℃培养箱内,将10μm荧光探针alp-0、2、5和6分别与细胞膜表面过量表达alp的hela细胞和alp低表达的正常组织细胞hek293于共聚焦培养皿内孵育1.5小时;或将hela细胞提前用碱性磷酸酶抑制剂p-bto预处理1小时,再与不同探针共同孵育1.5小时。随后弃去含有探针的培养基,并用 pbs缓冲液(ph=7.4)洗涤三次,去除未与膜表面建立共价连接的探针分子;加入1 ml无菌pbs缓冲液(ph=7.4),利用荧光显微镜进行细胞成像(λ
ex
=470
±
20nm,λ
em
=515nm)。
[0243]
实验结果:
[0244]
实验结果如图3所示,图3中,左侧7组为荧光探针alp-0、2、5和6在hela 细胞膜表面alp激活下对细胞膜的标记实验;右侧4组为荧光探针alp-0、2、5 和6对低表达alp的hek293细胞膜的标记实验。图中fl为绿色荧光信号; hoechst为hoechst 3342染色的细胞核信号;bf为白光照射下的细胞形态;merge 为以上几个视野的合成图;p-bto((-)-p-bromotetramisole oxalate)是一种商业化的 alp抑制剂.scale bar=50μm。
[0245]
由图3可知,具有共价键“锚点”的荧光探针alp-2、5、6与无标记能力的普通荧光探针alp-0相比,不仅能够实现对alp过表达细胞的识别,明显区分alp 低表达细胞株(hek293)与高表达细胞株(hela);而且能够利用alp水解探针后形成的qm活性中间体实现对阳性细胞胞膜的“锚定”。此外,在alp抑制剂组实验中,荧光探针alp-2、5和6对hela细胞标记的荧光信号减弱,这说明荧光探针标记作用的启动依赖于碱性磷酸酶特异性水解磷酸酯键的性质。再次,通过比较荧光探针alp-5和6对hela细胞标记的荧光半定量数据,可知荧光探针alp-6在更为接近生理环境的离体活细胞层面能够更有效地检测靶酶并标记阳性细
bromotetramisole oxalate)是一种商业化的alp抑制剂.scale bar=50μm.
[0255]
具有共价键“锚点”的增强型荧光探针alp-1、2与无标记能力的普通增强荧光探针alp nir-0相比,不仅能够实现对alp过表达细胞的识别,明显区分alp 低表达细胞株(hek293)与高表达细胞株(hela);而且能够利用alp水解探针后形成的qm活性中间体实现对阳性细胞的“锚定”。此外,在alp抑制剂组实验中,荧光探针alp nir-1和2对alp受抑制的hela细胞标记的荧光信号减弱,这说明荧光探针共价标记作用的启动依赖于alp特异性水解磷酸酯键而诱导产生qm 中间体的过程。再次,通过比较荧光探针alp nir-1和2对hela细胞标记的效果,可知具有多个潜在共价键“靶点”的增强型荧光探针alp nir-2在更为接近生理环境的离体活细胞层面能够更有效、精准地检测靶酶并标记阳性细胞。
[0256]
实验结论:多“锚点”增强型荧光探针alp nir-2能够在离体活细胞水平实现对alp过表达的肿瘤细胞的鉴别与精准定位作用,从而减少荧光探针在细胞成像实验中的弥散,降低背景信号,提高检测灵敏度。此外,结合前面的实验结果与结论,可知增强型荧光探针alp nir-1和2两者均具有潜在p-qm结构和高效的蛋白标记作用;此外,alp nir-2对alp的特异性检测能力也较为突出。综合以上结果,本发明具有多个潜在共价键“锚点”的增强型荧光探针alp nir-2不仅能够在细胞水平更为有效的实现对细胞膜表面蛋白共价标记作用,而且其在生物标记及生物医药领域具有更为可期的前景与实际应用价值。
[0257]
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1