一种制备异喹啉酮类化合物的方法与流程
1.本申请涉及药物化学领域,尤其涉及一种制备异喹啉酮类化合物的方法。
背景技术:
2.罗沙司他,化学名(4-羟基-1-甲基-7-苯氧基-异喹啉-3-羰基)-氨基]-乙酸,分子式为:c
19
h
16
n2o5,分子量为:352.11,cas号为:808118-40-3,化学结构式为:
[0003][0004]
罗沙司他是由珐博进(fibrogen)公司开发的一种治疗肾性贫血的疾病,于2017年11月在国内申请上市。该药是全球首个开发的小分子低氧诱导因子脯氨酰羟化酶抑制剂(hif-phi)类治疗肾性贫血的药物。低氧诱导因子(hif) 的生理作用不仅使红细胞生成素表达增加,也能使红细胞生成素受体以及促进铁吸收和循环的蛋白表达增加。罗沙司他通过模拟脯氨酰羟化酶(ph)的底物之一酮戊二酸来抑制ph酶,影响ph酶在维持hif生成和降解速率平衡方面的作用,从而达到纠正贫血的目的。罗沙司他为由慢性肾脏病引起的贫血患者提供了新的治疗手段。
[0005]
然而,在的现有罗沙司他的制备技术中,反应路线往往需要在低温、高温、密闭加压条件下反应,反应条件苛刻,对工艺对设备要求较高,且反应路线长,副反应多,导致后续纯化困难,使得合成的罗沙司他的产率和纯度低,此外,现有的合成方法需要昂贵的催化剂,从而不利于工业化生产。
[0006]
因此,需要开发一种具有路线合理、方便易行、适合工业化生产的异喹啉酮类化合物罗沙司他的合成方法。
[0007]
技术实现要素:
[0008]
本发明的目的在于提供一种具有路线合理、方便易行、产率和纯度高且适合工业化生产的式3结构的异喹啉酮类化合物的制备方法。
[0009]
本发明第一方面,提供一种制备式3化合物的方法,所述方法包括以下步骤:
[0010]
1)将式1化合物与酰氯反应得到式2化合物;
[0011][0012]
2)式2化合物与胺解试剂反应后经水解反应从而得到式3化合物,其中,所述的胺解试剂选自下组:甘氨酸、甘氨酸衍生物,或其组合;
[0013][0014]
其中,所述酰氯选自下组:r1c(o)cl、r2c(o)cl,或其组合;
[0015]
r1和r2各自独立地为c1-c10烷基或c6-c10芳基。
[0016]
在另一优选例中,r1和r2各自独立地为c1-c6烷基或c6-c10芳基。
[0017]
在另一优选例中,所述步骤1)中,得到的式2化合物为反应得到的含式2 化合物的混合物。
[0018]
在另一优选例中,所述步骤1)中,式1化合物在缚酸剂的作用下和在第一惰性溶剂中与酰氯反应,得到式2化合物。
[0019]
在另一优选例中,所述步骤2)中,式2化合物先经胺解试剂胺解后,经过第一碱试剂水解得到式3化合物。
[0020]
在另一优选例中,所述步骤1)中,所述的缚酸剂选自下组:三乙胺(tea)、 1,8-二氮杂二环十一碳-7-烯(dbu)、n,n-二异丙基乙胺(diea)、吡啶、n-甲基吗啉,或其组合。
[0021]
在另一优选例中,所述步骤1)中所述第一惰性溶剂选自下组:四氢呋喃、二氯甲烷、甲苯,或其组合。
[0022]
在另一优选例中,所述步骤1)中,所述酰氯选自下组:乙酰氯、三甲基乙酰氯、苯甲酰氯,或其组合。
[0023]
在另一优选例中,所述步骤1)中,所述酰氯和所述式1化合物的摩尔比为 1-4:1,较佳地2-3.5:1。
[0024]
在另一优选例中,所述步骤2)中,所述第一碱试剂选自下组:氢氧化钠、氢氧化钾、氢氧化锂,或其组合。
[0025]
在另一优选例中,所述步骤2)中,甘氨酸衍生物包括甘氨酸盐或甘氨酸酯。
[0026]
在另一优选例中,r1和r2各自独立地为甲基、乙基、正丙基、苯基、苄基、正丁基、异丁基或叔丁基。
[0027]
在另一优选例中,所述步骤2)中,所述胺解试剂和所述式1化合物的摩尔比为1-3:1。
[0028]
在另一优选例中,所述步骤1)中,所述的式1化合物与缚酸剂的摩尔比为1:1-6,较佳地1:2-4。
[0029]
在另一优选例中,所述步骤2)中,所述式2化合物与所述的第一碱试剂的摩尔比为1:1-6,较佳地1:2-3。
[0030]
在另一优选例中,所述步骤1)中,反应的温度为15-40℃,较佳地20-30℃。
[0031]
在另一优选例中,所述步骤1)中,反应的时间为0.5-24h,较佳地1-5h,更佳地3-5h。
[0032]
在另一优选例中,所述步骤2)中,反应的温度为15-40℃,较佳地20-30℃。
[0033]
在另一优选例中,所述步骤2)中,反应的时间为0.5-24h,较佳地4-8h。
[0034]
在另一优选例中,所述步骤2)中,所述胺解试剂和所述式2化合物的摩尔比为1-3:1。
[0035]
在另一优选例中,所述的的甘氨酸衍生物选自下组:甘氨酸钠、甘氨酸甲酯,或其组合。
[0036]
在另一优选例中,所述第一碱试剂包括氢氧化钠。
[0037]
在另一优选例中,所述甘氨酸盐包括甘氨酸钠。
[0038]
在另一优选例中,所述甘氨酸酯包括甘氨酸甲酯。
[0039]
在另一优选例中,所述步骤1)中,所述缚酸剂与所述式1化合物的摩尔比为1-5:1。
[0040]
在另一优选例中,所述步骤1)中,式1化合物、缚酸剂和第一惰性溶剂混合后,降温至0-10℃,加入酰氯,升温至20-30℃,反应得到式2化合物。
[0041]
在另一优选例中,所述步骤1)中,式1化合物、缚酸剂和第一惰性溶剂混合后,降温至0-10℃,加入酰氯,升温至20-30℃,反应得到含式2化合物的混合物,任选地,含式2化合物的混合物经后处理得到式2化合物。
[0042]
在另一优选例中,所述步骤1)中,将式2化合物降温至0-10℃,加入胺解试剂,升温至20-40℃(较佳地20-30℃)进行胺解反应(优选地,反应时间为 1-4h,较佳地2-3h),检测胺解反应完成后,加入第一碱试剂,进行水解反应 (优选地,反应时间为1-4h,较佳地1-3h),得到化合物3。
[0043]
在另一优选例中,所述步骤1)中,将式2化合物降温至0-10℃,加入胺解试剂,升温至20-40℃(较佳地20-30℃)进行胺解反应(优选地,反应时间为 1-4h,较佳地2-3h),检测胺解反应完成后,加入第一碱试剂,进行水解反应 (优选地,反应时间为1-4h,较佳地1-3h),得到的反应液经二氯甲烷和水萃取,水相用稀盐酸调ph至2-3,析出固体,过滤,并用丙酮洗涤,得到化合物 3。
[0044]
在另一优选例中,所述步骤1)中得到的含式2化合物的混合物无需后处理即可按照所述步骤2)进行后续反应。
[0045]
在另一优选例中,所述的反应在常压下进行。
[0046]
在另一优选例中,所述步骤1)中,所述的反应在常压下进行。
[0047]
在另一优选例中,所述步骤2)中,所述的反应在常压下进行。
[0048]
在另一优选例中,所述式1化合物通过以下方法制备:
[0049]
(a)将式s1化合物在第二惰性溶剂中与卤代试剂反应得到式s2化合物;
[0050]
(b)将式s2化合物在钯催化剂和第二碱试剂的存在下,在第三惰性溶剂中与甲基
化试剂反应得到式1化合物。
[0051][0052]
其中x为cl、br或i。
[0053]
在另一优选例中,所述步骤(a)中,所述的反应在常压下进行。
[0054]
在另一优选例中,所述步骤(b)中,所述的反应在常压下进行。
[0055]
在另一优选例中,所述步骤a)中,所述第二惰性溶剂选自下组:乙腈、甲醇、乙醇、乙酸乙酯、二氯甲烷中,或其组合。
[0056]
在另一优选例中,所述步骤a)中,所述卤代试剂选用下组:ncs、nbs、 nis、二氯海因、二溴海因、二碘海因、溴素、单质碘,或其组合。
[0057]
在另一优选例中,所述步骤a)中,式s1化合物和卤代试剂的摩尔比为1: 1-3。
[0058]
在另一优选例中,所述步骤b)中,所述甲基化试剂选自下组:三甲基硼、甲基硼酸、甲基硼酸异丙酯、甲基三氟硼酸钾,或其组合。
[0059]
在另一优选例中,所述步骤b)中,所述第二碱试剂选自下组:naoh、koh、 lioh、na2co3、k2co3、na3po4、k3po4,或其组合。
[0060]
在另一优选例中,所述步骤b)中,所述钯催化剂选自下组:醋酸钯、二(三苯基膦)二氯化钯、四(三苯基膦)钯、三(苄亚基丙酮)二钯、双(二苯基膦)二茂铁二氯化钯、三苯基膦二氯化钯,或其组合。
[0061]
在另一优选例中,所述步骤b)中,所述第三惰性溶剂包括乙二醇甲醚和水混合液。
[0062]
在另一优选例中,所述步骤b)中,所述第三惰性溶剂包括乙二醇甲醚和水混合液,乙二醇甲醚和水的体积比为2-8:1,较佳地2-5:1。
[0063]
在另一优选例中,所述步骤b)中,所述的式s2化合物与甲基化试剂的摩尔比为1:0.5-4,较佳地1:1-2。
[0064]
在另一优选例中,所述步骤b)中,所述的式s2化合物与第二碱试剂的摩尔比为1:0.5-5,较佳地1:1-3。
[0065]
在另一优选例中,所述步骤a)中,所述第二惰性溶剂为乙腈、二氯甲烷或乙腈和二氯甲烷的混合溶液。
[0066]
在另一优选例中,乙腈与二氯甲烷的体积比为1-2:1。
[0067]
在另一优选例中,所述步骤a)中,所述第三惰性溶剂包括乙二醇甲醚和水混合液,且乙二醇甲醚与水的体积比为
[0068]
在另一优选例中,所述步骤a)中,式s1化合物加入到第二惰性溶剂中,降温至0-10℃,加入卤代试剂后,升温至室温反应,得到式s2化合物。
[0069]
在另一优选例中,所述步骤a)中,式s1化合物加入到第二惰性溶剂中,降温至0-10℃,加入卤代试剂后,升温至室温反应,检测反应完成后,浓缩至干,用乙腈打浆,过滤,得到式s2化合物。
[0070]
在另一优选例中,所述步骤b)中,式s2化合物、钯催化剂、第二碱试剂、甲基化试剂
和第三惰性溶剂中混合后,升温至90-100℃反应(优选地,反应的时间为3-5h),得到得到式1化合物。
[0071]
在另一优选例中,所述步骤b)中,式s2化合物、钯催化剂、第二碱试剂、甲基化试剂和第三惰性溶剂中混合后,升温至90-100℃反应(优选地,反应的时间为3-5h),检测反应完成后,冷却至20-30℃,再加入纯水,加盐酸调节ph 至2-3过滤,用甲醇洗涤得到得到式1化合物。
[0072]
在另一优选例中,所述步骤a)中,所述卤代试剂选自下组:nbs、ncs、 nis、二碘海因,或其组合。
[0073]
在另一优选例中,所述步骤a)中,化合物s1和卤代试剂的摩尔比为1: 1.05-1.3。
[0074]
在另一优选例中,所述步骤b)中,所述甲基化试剂选自下组:三甲基硼、甲基硼酸、甲基硼酸异丙酯,或其组合。
[0075]
在另一优选例中,所述步骤b)中,所述的第二碱试剂包括k3po4。
[0076]
在另一优选例中,所述步骤b)中,所述的钯催化剂包括二(三苯基膦)二氯化钯。
[0077]
在另一优选例中,所述的反应无需密闭环境条件下反应。
[0078]
在另一优选例中,所述的反应在密闭和开放条件下反应。
[0079]
在另一优选例中,所述步骤(a)中,反应的温度为室温。
[0080]
在另一优选例中,所述步骤(a)中,反应的时间为0.5-24h,较佳地2-6h。
[0081]
在另一优选例中,所述步骤(b)中,反应的温度为70-120℃,较佳地 90-100℃。
[0082]
在另一优选例中,所述步骤(b)中,反应的时间为0.5-24h,较佳地2-5h。
[0083]
本发明第二方面,提一种异喹啉酮类化合物中间体,所述异喹啉酮类化合物中间体的结构如式2所示。
[0084][0085]
其中,r1和r2各自独立地为c1-c10烷基或c6-c10芳基。
[0086]
在另一优选例中,所述异喹啉酮类化合物中间体为:
[0087][0088]
本发明第三方面,提供一种制备如本发明第二方面所述的中间体的方法,所述的方法包括步骤:
[0089]
1)将式1化合物与酰氯反应得到式2化合物;
[0090][0091]
其中,r1和r2如本发明第一方面所定义。
[0092]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例) 中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
[0093]
本发明经过广泛而又深入的研究,意外地发现一种制备式3结构化合物的方法。本发明所述的式3结构化合物的制备方法具有路线合理、方便易行、制备的产率和纯度高、适合工业化生产等优势。在此基础上,发明人完成了本发明。
[0094]
术语
[0095]
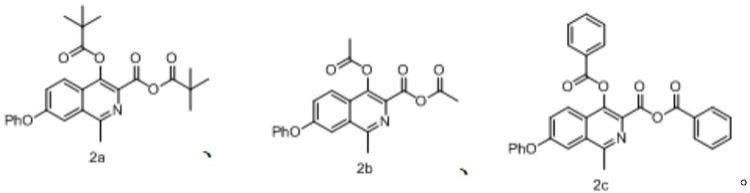
如本文所用,术语“包含”、“包括”、“含有”可互换使用,不仅包括封闭式定义,还包括半封闭、和开放式的定义。换言之,所述术语包括了“由
……
构成”、“基本上由
……
构成”。
[0096]
如本文所用,术语“烷基”指只含碳原子的直链(即,无支链)或支链饱和烃基,或直链和支链组合的基团。当烷基前具有碳原子数限定(如c1-c10烷基) 指所述的烷基含有1-10个碳原子,代表性实例包括但不限于甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、或类似基团。
[0097]
术语“芳基”是指芳香环状烃类化合物基团,例如具有1、或2个环,尤其指单环和双环基团,如苯基、联苯基或萘基。凡含有两个或两个以上芳香环(双环等),芳基基团的芳香环可由单键联接(如联苯),或稠合(如萘、蒽等等)。当芳基前有碳原子数限定时,指的是芳基的环碳原子个数,例如c6-c10芳基指的是具有6-10个环碳原子的芳基,代表性实例包括但不限于苯基、联苯基或萘基。
[0098]
在本发明中,除非另有说明书,所有的取代基为未取代的取代基。
[0099]
如本文所用,如本文所用与结构可互换使用。
[0100]
本发明中使用的缩写形式及其含义如下表所述:
[0101][0102][0103]
如本文所使用“惰性溶剂”是指不与反应中的其它物质(如原料、催化剂等) 发生反应的溶剂。
[0104]
如本文所用与结构可互换使用。
[0105]
制备方法
[0106]
式3结构的异喹啉酮类化合物的制备方法
[0107]
本发明提供一种式3结构化合物的制备方法,具体地,所述方法包括以下步骤:
[0108]
1)将式1化合物与酰氯反应得到式2化合物;
[0109]
[0110]
2)式2化合物与胺解试剂反应后经水解反应从而得到式3化合物,其中,所述的胺解试剂选自下组:甘氨酸、甘氨酸衍生物,或其组合;
[0111][0112]
其中,所述酰氯选自下组:r1c(o)cl、r2c(o)cl,或其组合;
[0113]
r1和r2各自独立地为c1-c10烷基或c6-c10芳基。
[0114]
在本发明的一个优选例中,所述步骤1)中,式1化合物在缚酸剂的作用下和在第一惰性溶剂中与酰氯反应,得到式2化合物。
[0115]
在本发明的另一优选例中,所述步骤2)中,式2化合物先经胺解试剂胺解后,经过第一碱试剂水解得到式3化合物。
[0116]
在另一优选例中,所述步骤1)中,所述的缚酸剂包括(但不限于):三乙胺(tea)、1,8-二氮杂二环十一碳-7-烯(dbu)、n,n-二异丙基乙胺(diea)、吡啶、 n-甲基吗啉,或其组合。
[0117]
在另一优选例中,所述步骤1)中所述第一惰性溶剂包括(但不限于):四氢呋喃、二氯甲烷、甲苯,或其组合。
[0118]
在另一优选例中,所述步骤1)中,所述酰氯包括(但不限于):乙酰氯、三甲基乙酰氯、苯甲酰氯,或其组合。
[0119]
在另一优选例中,所述步骤1)中,所述酰氯和所述式1化合物的摩尔比为 1-4:1,较佳地2-3.5:1。
[0120]
在另一优选例中,所述步骤2)中,所述第一碱试剂包括(但不限于):氢氧化钠、氢氧化钾、氢氧化锂,或其组合。
[0121]
在另一优选例中,所述步骤2)中,甘氨酸衍生物包括甘氨酸盐或甘氨酸酯。
[0122]
在另一优选例中,r1和r2各自独立地为甲基、乙基、正丙基、苯基、苄基、正丁基、异丁基或叔丁基。
[0123]
在另一优选例中,所述步骤2)中,所述胺解试剂和所述式1化合物的摩尔比为1-3:1。
[0124]
在另一优选例中,所述步骤1)中,所述的式1化合物与缚酸剂的摩尔比为 1:1-6,较佳地1:2-4。
[0125]
在另一优选例中,所述步骤2)中,所述式2化合物与所述的第一碱试剂的摩尔比为1:1-6,较佳地1:2-3。
[0126]
在另一优选例中,所述步骤1)中,反应的温度为15-40℃,较佳地20-30℃。
[0127]
在另一优选例中,所述步骤2)中,反应的温度为15-40℃,较佳地20-30℃。
[0128]
在另一优选例中,所述的的甘氨酸衍生物包括(但不限于):甘氨酸钠、甘氨酸甲酯,或其组合。
[0129]
在另一优选例中,所述第一碱试剂包括氢氧化钠。
[0130]
在另一优选例中,所述甘氨酸盐包括甘氨酸钠。
[0131]
在另一优选例中,所述甘氨酸酯包括甘氨酸甲酯。
[0132]
在另一优选例中,所述步骤1)中,所述的反应在常压下进行。
[0133]
在另一优选例中,所述步骤2)中,所述的反应在常压下进行。
[0134]
在本发明的另一优选例中,所述式1化合物通过以下方法制备:
[0135]
(a)将式s1化合物在第二惰性溶剂中与卤代试剂反应得到式s2化合物;
[0136]
(b)将式s2化合物在钯催化剂和第二碱试剂的存在下,在第三惰性溶剂中与甲基化试剂反应得到式1化合物。
[0137][0138]
其中x为cl、br或i。
[0139]
在另一优选例中,所述步骤(a)中,所述的反应在常压下进行。
[0140]
在另一优选例中,所述步骤(b)中,所述的反应在常压下进行。
[0141]
在另一优选例中,所述步骤a)中,所述第二惰性溶剂包括(但不限于):乙腈、甲醇、乙醇、乙酸乙酯、二氯甲烷中,或其组合。
[0142]
在另一优选例中,所述步骤a)中,所述卤代试剂选用下组:ncs、nbs、 nis、二氯海因、二溴海因、二碘海因、溴素、单质碘,或其组合。
[0143]
在另一优选例中,所述步骤a)中,式s1化合物和卤代试剂的摩尔比为1: 1-3。
[0144]
在另一优选例中,所述步骤b)中,所述甲基化试剂包括(但不限于):三甲基硼、甲基硼酸、甲基硼酸异丙酯、甲基三氟硼酸钾,或其组合。
[0145]
在另一优选例中,所述步骤b)中,所述第二碱试剂包括(但不限于):naoh、koh、lioh、na2co3、k2co3、na3po4、k3po4,或其组合。
[0146]
在另一优选例中,所述步骤b)中,所述钯催化剂包括(但不限于):醋酸钯、二(三苯基膦)二氯化钯、四(三苯基膦)钯、三(苄亚基丙酮)二钯、双(二苯基膦)二茂铁二氯化钯、三苯基膦二氯化钯,或其组合。
[0147]
在另一优选例中,所述步骤(a)中,反应的温度为室温。
[0148]
在另一优选例中,所述步骤(a)中,反应的时间为0.5-24h,较佳地2-6h。
[0149]
在另一优选例中,所述步骤(b)中,反应的温度为70-120℃,较佳地 90-100℃。
[0150]
在另一优选例中,所述步骤(b)中,反应的时间为0.5-24h,较佳地2-5h。
[0151]
中间体
[0152]
本发明还提供一种异喹啉酮类化合物中间体,所述异喹啉酮类化合物中间体的结构如式2所示:
[0153][0154]
其中,r1和r2各自独立地为c1-c10烷基或c6-c10芳基。
[0155]
在另一优选例中,所述异喹啉酮类化合物中间体为:
[0156][0157]
本发明还提供一种制备异喹啉酮类化合物中间体的方法,所述的方法包括步骤:
[0158]
1)将式1化合物与酰氯反应得到式2化合物;
[0159][0160]
其中,r1和r2如上所定义。
[0161]
本发明的主要优点包括:
[0162]
1)本发明意外地采用“混合酸酐法”来构建异喹啉酮类化合物化学结构中的酰胺键,使式1化合物在缚酸剂的作用下与酰氯反应,得到式2化合物酸酐,然后再“一锅法”与甘氨酸或其衍生物胺解,之后可以使用碱水解分子结构中4位的酯基,从而非常方便地得到式3化合物,且避免使用如dcc、edc等常规缩合剂,消除了反应中产生缩合剂副产物。
[0163]
2)本发明中,由于将化合物2胺解制备化合物3的过程中使用的溶剂或试剂可与制备化合物2相同,不需要进行任何后处理,可以实现化合物1到异喹啉酮类化合物“一锅法”连投,这样大大缩短了生成周期,提高生产效率,更加利于工业化放大生产。
[0164]
3)本发明的式3化合物的制备工艺相较于现有技术,制备过程简单,反应时间短,产率高,副产物少,纯度高,具有较好的工业化前景。
[0165]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
[0166]
实施例
[0167]
在实施例1-6中,所有的反应均在常压(标准大气压)条件下进行,且室温指的是25
±
5℃。
[0168]
实施例1
[0169][0170]
将4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(10g,33.87mmol)加入二氯甲烷中,降温至0-10℃之后,分批加入n-溴代琥珀酰亚胺(nbs)固体(35.6mmol),加完后升温至室温搅拌反应4-5小时,tlc板检测反应完成,浓缩至干,用乙腈打浆,过滤,得到4-羟基-1-溴-7-苯氧基异喹啉-3-甲酸甲酯11.85g,收率93.9%。
[0171]
将上述4-羟基-1-溴-7-苯氧基异喹啉-3-甲酸甲酯(7.0g,0.019mol),四(三苯基膦)钯(0.05eq),甲基硼酸(1.5eq),磷酸钾(2.0eq)依次加入乙二醇甲醚140ml和水28ml,升温至90-100℃反应3h,tlc检测反应完成,冷却至 20-30℃,再加入纯水,加盐酸调节ph至2-3,过滤,用甲醇淋洗得4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酸5.1g,收率91%。
[0172]
实施例2
[0173][0174]
将4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(10g,33.87mmol)加入乙腈中,降温至0-10℃之后,分批加入n-氯代琥珀酰亚胺(ncs)固体(71.2mmol),加完后升温至室温搅拌反应3.5-4.5小时,tlc板检测反应完成,浓缩至干,用乙腈打浆,过滤,得到4-羟基-1-氯-7-苯氧基异喹啉-3-甲酸甲酯10.4g,收率92%。
[0175]
将上述4-羟基-1-氯-7-苯氧基异喹啉-3-甲酸甲酯(6.2g,0.019mol),三苯基膦二氯化钯(0.05eq),三甲基硼(1.5eq),na3po4(2.0eq)依次加入乙二醇甲醚124ml和水24.8ml,升温至90-100℃反应4h,tlc检测反应完成,冷却至 20-30℃,再加入纯水,加盐酸调节ph至2-3,过滤,用甲醇淋洗得4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酸4.9g,收率88%。
[0176]
实施例3
[0177][0178]
将4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(10g,33.87mmol)加入乙腈100ml 和二氯甲烷100ml,降温至0-10℃之后,分批加入二碘海因(90mmol),加完后升温至室温搅拌反应4-4.5小时,tlc板检测反应完成,浓缩至干,用乙腈打浆,过滤,得到4-羟基-1-碘-7-苯氧基异喹啉-3-甲酸甲酯11.45g,收率90.7%。
[0179]
将上述4-羟基-1-碘-7-苯氧基异喹啉-3-甲酸甲酯(7.0g,0.019mol),三苯基膦二氯化钯(0.05eq),甲基硼酸异丙酯(1.5eq),k2co3(2.0eq)依次加入乙二醇甲醚105ml和水
35ml,升温至90-100℃反应4h,tlc检测反应完成,冷却至20-30℃,再加入纯水,加盐酸调节ph至2-3,过滤,用甲醇淋洗得4-羟基
ꢀ-
1-甲基-7-苯氧基异喹啉-3-甲酸5.3g,收率95.4%。
[0180]
实施例4
[0181][0182]
将4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酸(2.0g,6.77mmol)加入20ml四氢呋喃中,再加入n,n-二异丙基乙胺(3.0eq),降温至0-10℃,缓慢加入三甲基乙酰氯(2.2eq),加完升温至20-30℃反应2-3小时,tlc检测反应完成,得到含有新戊酸-4-新戊酰氧基-1-甲基-7-苯氧基异喹啉-3-甲酸酐的混合物,所得混合物直接进行下一步反应,其中取少量混合物浓缩过滤,取滤渣进行柱层析后得到式2a化合物:
[0183]
式2a化合物ms m/z(esi):464(m+1);1h nmr(400mhz,cdcl3)δ7.94 (dd,j=8.5,1.1hz,1h),7.51(s,1h),7.50
–
7.40(m,3h),7.23(d,j=7.4hz,1h), 7.10(dd,j=8.5,0.8hz,2h),2.77(s,3h),1.52(s,9h),1.41(s,9h).
[0184]
上述得到混合物降温至0-10℃,缓慢加入甘氨酸钠(2.0eq,以式1化合物计),升温至20-30℃反应2-3h,tlc检测反应完成。然后加入naoh(2eq,以式1化合物计),室温搅拌反应2h。二氯甲烷和水萃取,水相用稀盐酸调ph至2-3,析出固体,过滤,并用丙酮淋洗,烘干即得式3化合物产品2.15g,收率90%。式3化合物ms m/z(esi):353(m+1);1h nmr(400mhz,dmso)δ13.07(d,j= 196.2hz,2h),9.10(t,j=5.9hz,1h),8.30(d,j=9.0hz,1h),7.62(d,j=2.3hz, 1h),7.51(ddd,j=15.9,8.6,5.0hz,3h),7.26(t,j=7.4hz,1h),7.19(d,j=7.7 hz,2h),4.06(d,j=6.1hz,2h),2.71(s,3h).
[0185]
实施例5
[0186][0187]
将4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酸(2.0g,6.77mmol)加入15ml二氯甲烷中,再加入三乙胺(4.0eq),降温至0-10℃,缓慢加入乙酰氯(3.2eq),加完升温至20-30℃反应1.5-2.5小时,tlc检测反应完成,得到含有乙酸-4-乙酰氧基-1-甲基-7-苯氧基异喹啉-3-甲酸酐的混合物,所得混合物直接进行下一步反应,其中取少量混合物浓缩过滤,取滤渣进行柱层析后得到式2b化合物::
[0188]
式2b化合物ms m/z(esi):380(m+1)。1h nmr(400mhz,dmso)δ7.95 (dd,j=8.5,1.1hz,1h),7.53(s,1h),7.51
–
7.41(m,3h),7.24(d,j=7.4hz,1h), 7.11(dd,j=8.5,0.8hz,2h),2.77(s,3h),2.23(s,3h),2.19(s,3h).
[0189]
上述得到混合物降温至0-10℃,缓慢加入甘氨酸甲酯(2.0eq,以式1化合物计),升温至30℃反应3h,tlc检测反应完成。然后加入koh(2eq,以式1 化合物计),室温搅拌反应2h。二氯甲烷和水萃取,水相用稀盐酸调ph至2-3,析出固体,过滤,并用丙酮淋洗,烘干即得
式3化合物产品2.19g,收率92%。式3化合物的分析数据与实施例4相同。
[0190]
实施例6
[0191][0192]
将4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酸(2.0g,6.77mmol)加入20ml甲苯中,再加入dbu(2.0eq),降温至0-10℃,缓慢加入苯甲酰氯(2.8eq),加完升温至20-30℃反应2.5-3.5小时,tlc检测反应完成,得到含有苯甲酸-4-苯甲酰氧基-1-甲基-7-苯氧基异喹啉-3-甲酸酐的混合物,所得混合物直接进行下一步反应,其中取少量混合物浓缩过滤,取滤渣进行柱层析后得到式2c化合物:
[0193]
式2c化合物ms m/z(esi):504(m+1);1h nmr(400mhz,dmso)δ8.19(dd, j=8.5,0.8hz,4h),8.03(dd,j=8.5,1.1hz,1h),7.82
–
7.78(m,2h),7.73
–
7.67 (m,3h),7.63
–
7.59(m,2h),7.53
–
7.43(m,3h),7.21(d,j=7.4hz,1h),7.10 (dd,j=8.5,0.8hz,2h),2.78(s,3h).
[0194]
上述得到混合物降温至0-10℃,缓慢加入甘氨酸(2.0eq,以式1化合物计),升温至30℃反应3h,tlc检测反应完成。然后加入lioh(3eq,以式1化合物计),室温搅拌反应2h。二氯甲烷和水萃取,水相用稀盐酸调ph至2-3,析出固体,过滤,并用丙酮淋洗,烘干即得式3化合物产品2.24g,收率94%。式3化合物的分析数据与实施例4相同。
[0195]
对比例
[0196]
按照cn104024227公开了罗沙司他的合成方法,具体路线如下:
[0197][0198]
该路线的制备的罗沙司他总收率为49.2%,收率较低,在实际生产中应用受限制。
[0199]
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1