异噁唑类FXR受体激动剂、其制备方法和医药用途
本发明属于药物化学领域,具体涉及异噁唑类fxr受体激动剂、其制备方法和医药用途。
背景技术:
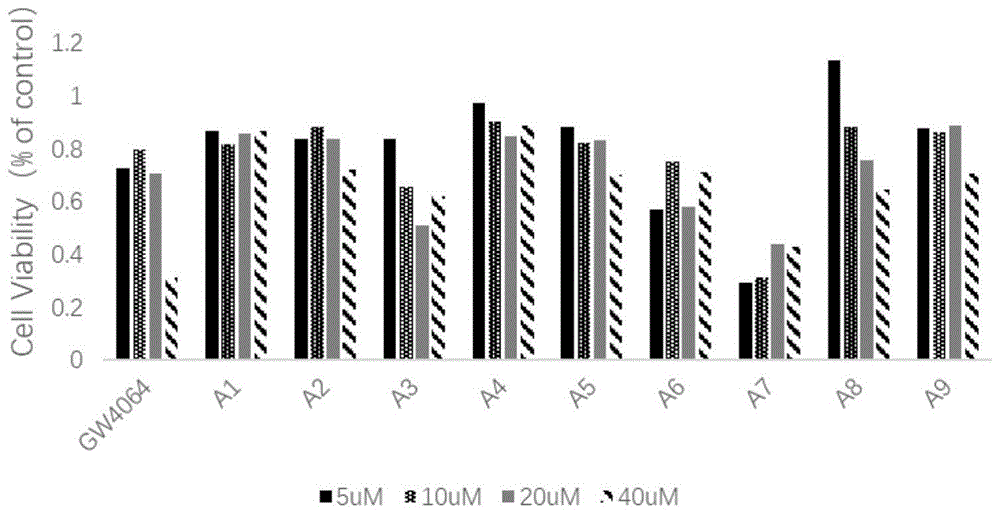
:血脂异常是指血浆中的总胆固醇(totalcholesterol,tc)、甘油三酯(triglyceride,tg)和低密度脂蛋白胆固醇(low-densitylipidcholesterol,ldl-c)增多,高密度脂蛋白胆固醇(high-densitylipidcholesterol,hdl-c)减少。ldl可被氧化成oxldl,过多的oxldl在巨噬细胞内堆积变成泡沫细胞,同时引起血管内皮细胞凋亡,引起炎性反应,共同促进动脉粥样硬化斑块形成;新生的小颗粒hdl接受从血管壁巨噬细胞中流出的胆固醇,经酯化后转运到肝脏并代谢成胆酸盐清除出体外,完成胆固醇逆转运(reversecholesteroltransport,rct)。高tg引起过度脂交换,会形成含胆固醇少、tg多的大颗粒hdl,使rct能力强的小颗粒hdl-c水平下降,胆固醇在血管内壁沉积,增加了动脉粥样硬化法尼醇x受体(farnesoidxreceptor,fxr),属于核受体超家族的一员。fxr参与调节机体的多种生理活动,入胆汁酸代谢、脂代谢、糖代谢和肝脏保护。基于其在胆汁酸代谢和脂代谢方面的作用,fxr被认为有望成为治疗血脂异常的新靶点。fxr激活后,诱导肠道细胞合成及分泌人成纤维细胞生长因子(fgf)19,fgf19入肝后激活肝细胞表面的fgf受体4,进而抑制胆汁酸合成限速酶胆固醇7羟化酶1的表达,减少胆汁酸的合成。激活fxr能促进b类i型清道夫受体的表达,抑制固醇调节元件结合蛋白1c表达,诱导过氧化物酶体增殖物激活受体α表达,促进胆固醇逆向转运入肝,减少脂质合成,促进脂肪酸β氧化。在有关异噁唑类fxr激动剂的研究中,maloney等筛选获得了第一个非甾体fxr激动剂gw4064(14),ec50值为15nmol/l。但由于其生物利用度低,且结构中的二苯乙烯基团存在潜在的毒性,因此限制了其进行进一步的研究,目前gw4064主要用作fxr研究中的工具药物。为了克服gw4064的缺陷,后续许多制药企业和研究人员合成了多个gw4064的衍生物。以fxr为靶点,开展异噁唑类衍生物调血脂药物的研究,对降低残留的心血管风险具有重要意义。技术实现要素:本发明的目的是提供一种异噁唑类fxr受体激动剂。本发明的另一目的是提供该异噁唑类fxr受体激动剂的制备方法。本发明的又一目的是提供该化合物的医药用途。式(i)所示的化合物、其药学上可接受的盐、同位素标记物:其中r1,r2分别独立的选自h,conh(ch2)nch3,coo(ch2)nch3,o(ch2)nch3,cl,br,f,i,cooh,no2,so3h,cn,n代表0、1、2、3、4、5、6。所述的式(i)所示的化合物、其药学上可接受的盐、同位素标记物,优选r1,r2分别独立的选自表h,conh(ch2)nch3,coo(ch2)nch3,o(ch2)nch3;n优选代表0、1、2。所述的式(i)所示的化合物进一步优选选自以下任意一种:编号r1、r2a1-h、-ha2-h、-conhch3a3-conhch3、-ha4-cooch3、-ha5-cooc2h5、-ha6-h、-cooch3a7-h、-cooc2h5a8-och3、-ha9-h、-och3。所述的式(i)所示的化合物的制备方法,反应路线如下:(a)nh2oh·hcl,naoh,etoh;(b)ncs,dmf;(c)naoch3,ch3oh,thf;(d)lialh4,thf;(e)cbr4,pph3,dcm,n2;(f)n-boc-4-羟基哌啶,18-冠醚-6,(ch3)3cok,thf,n2;(g)tfa,thf;(h-i)bis(trichloromethyl)carbonate,et3n,dcm.所述的式(i)所示的化合物、其药学上可接受的盐、同位素标记物在制备fxr受体激动剂中的应用。所述的式(i)所示的化合物、其药学上可接受的盐、同位素标记物在制备治疗或预防高血脂症、动脉粥样硬化、非酒精性脂肪肝炎、ⅱ型糖尿病糖尿病的药物中的用途。药理实验及实验实施例中化合物的代号等同于以上代号所对应的化合物结构。有益效果:本发明化合物的活性结果可以看出,本课题合成的化合物在40um下对3t3-l1细胞均有抑制活性(见图1)。在对hepg2肝癌细胞内fxr和fxr下游基因mrna表达的影响中,化合物a4能上调fxrmrna,shpmrna和bsepmrna的转录水平,下调bsrebp-1cmrna的转录水平(见图2-图5)。在fxr激动活性测试中,10μmol/l的浓度下化合物a4能明显激活fxr(见图6)。本发明化合物用于调血脂、降低心血管疾病的作用机制、作用强度和作用时间还需要在进一步的研究中阐述。附图说明图1化合物对3t3-l1细胞增殖活性的影响图2化合物对fxrmrna转录的影响图3化合物对shpmrna转录的影响图4化合物对bsepmrna转录的影响图5化合物对bsrebp-1cmrna转录的影响图6化合物a4对fxr的激动活性具体实施方式本发明的权利要求书特别陈述了本发明的新特征。在下文的陈述了利用本发明原理的示例性实施方式。通过参考以下内容可以更好地理解本发明的特征和优点。尽管本文描述了本申请的优选实施方式,但是这些实施方式仅作为示例提供。应该理解本文所述的本申请实施方式的变体也可用于实施本申请的技术方案。本领域普通技术人员应理解,可出现多种变体、变化和替换而不脱离本申请的范围。应理解本申请各个方面的保护范围由权利要求书决定,并且这些权利要求范围内的方法和结构以及其等价的方法和结构均在本申请权利要求书涵盖的范围之内。部分化合物的制备如下:熔点用xrc-1显微熔点仪测定(温度计未经校正),ir用nicoletis10型傅立叶变换红外光谱仪测定(kbr压片),1h-nmr核磁共振由brukerav300型(300mhz)核磁共振仪测定(tms为内标),质谱分别由agilentq-tof6520(hr-ms)、agilent1100lc-msd-trap/sl型质谱仪(esi-ms)测定。柱层析硅胶为100-200目或200-300目硅胶(青岛海洋化工厂),洗脱剂为石油醚-乙酸乙酯体系或二氯甲烷-甲醇体系。薄层层析(tlc)用gf254薄层层析板(烟台江友硅胶开发有限公司);tlc展开体系为石油醚-乙酸乙酯系统或二氯甲烷-甲醇系统,必要时加入少量乙酸或三乙胺;tlc在zf7型三用紫外分析仪(河南巩义予华仪器有限公司)下照射显示。实施例1中间体2-三氟甲氧基苯甲醛肟(2)的合成冰浴下,将h2noh·hcl(2.2g,31.7mmol)和naoh(1.2g,30.0mmol)溶于h2o中,配制成溶液a。将2-三氟甲氧基苯甲醛(5g,26.3mmol)溶于ch3ch2oh中,将配制好的溶液a逐滴加入,室温下反应1h。减压除去ch3ch2oh,加入h2o稀释,用乙酸乙酯(ea)和h2o萃取分液,饱和食盐水除水,无水na2so4干燥,减压旋干,直接下投。粗品5g,产率:93%。实施例2中间体2-三氟甲氧基-n-羟基-氯代苯甲醛肟(3)的合成将2-三氟甲氧基苯甲醛肟(粗品5g,24.4mmol)溶于dmf中,25℃以下将n-氯代丁二酰亚胺(ncs)(2.5g,18.7mmol)分批加入其中,室温下反应3h。反应液用ea和h2o萃取,有机相水洗,饱和食盐水除水,无水na2so4干燥,减压旋干,直接下投。粗品4.5g,产率77.1%。实施例3中间体3-(2-三氟甲氧基苯基)-5-环丙基异噁唑-4-甲酸甲酯(4)的合成冰盐浴下,将naoch3(2.64g,48.9mmol)溶于ch3oh中,配制成溶液b。将3-环丙基-3-氧代丙酸甲酯(5.34g,37.6mmol)溶于ch3oh中。将溶液b缓慢加入其中,加入的时间大于5min。将2-三氟甲氧基-n-羟基-氯代苯甲醛肟(4.5g,18.8mmol)溶于ch3oh中,冰盐浴下逐滴加入到上面混合液中,反应温度应始终小于-5℃。5min后,将冰盐浴撤去,升温至35℃,反应12h。用ch3cooh中和过量的naoch3,减压除去ch3oh,用ea和h2o萃取。饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化,产物2.5g,产率:40.7%。1hnmr(dmso-d6,400mhz)δ7.65-7.60(m,1h,ph),7.53(dd,j=7.6,2hz,1h,ph),7.48-7.43(m,2h,ph),3.68(s,3h,-cooch3),2.89(m,j=6.8hz,1h,-ch(ch2)2),1.30(m,j=6.8hz,4h,-ch(ch2)2)。实施例4中间体3-(2-三氟甲氧基苯基)-4-羟甲基-5-环丙基异噁唑(5)的合成将lialh4(0.58g,15.3mmol)置于茄型瓶中,冰浴下,缓慢滴入无水thf,搅拌,混合均匀。待气泡减少后,将3-(2-三氟甲氧基苯基)-5-环丙基异噁唑-4-甲酸甲酯(2.5g,7.6mmol)溶于无水thf中,缓慢滴入,反应1h。缓慢滴入饱和na2so4溶液终止反应,加入15%naoh溶液沉淀铝离子,用硅藻土过滤,ea洗涤滤饼。减压除去含产物的混合液,用ea和h2o萃取,有机相用饱和食盐水除水,无水na2so4干燥,减压旋干,直接下投。产率:2.1g,产率:91.9%。msm/z[m+h]+=300.1.实施例5中间体3-(2-三氟甲氧基苯基)-4-溴甲基-5-环丙基异噁唑(6)的合成冰盐浴下,将3-(2-三氟甲氧基苯基)-4-羟甲基-5-环丙基异噁唑(1g,3.34mmol)和pph3(1.31g,5.0mmol)溶于无水dcm中,搅拌至完全溶解。n2保护下,缓慢加入cbr4(1.66gcbr4,5.0mmol)的无水dcm溶液,反应1h。反应液直接减压旋干,柱层析纯化。产物:0.6g,产率:49.5%。1hnmr(cdcl3,400mhz)δ7.55-7.50(m,1h,ph),7.49-7.45(m,1h,ph),7.40-7.32(m,2h,ph),4.27(s,2h,ar-ch2-br),2.11-1.99(m,1h,-ch(ch2)2),1.24-1.17(m,2h,-ch(ch2)2),1.17-1.09(m,2h,-ch(ch2)2).实施例6中间体3-(2-三氟甲氧基苯基)-4-(4-氧甲基-n-boc-哌啶)-5-环丙基异噁唑(7)的合成在三颈瓶中加入n-boc-4-羟基哌啶(367mg,1.82mmol)、18-冠醚-6(482mg,1.82mmol),用无水thf溶解。将叔丁醇钾(204.2mg,1.82mmol)溶于无水thf中,混合均匀,逐滴加入至三颈瓶中。n2保护下,搅拌1h。将3-(2-三氟甲氧基苯基)-4-溴甲基-5-环丙基异噁唑(600mg,1.66mmol)溶于无水thf中,逐滴加入,搅拌过夜。用ch3cooh中和过量的叔丁醇钾,用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。产物:401mg,产率:50.0%。1hnmr(dmso-d6,300mhz)δ7.92-7.61(m,4h,ph),4.30(s,2h,ar-ch2-o-),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.66(m,4h,-o-ch2-ch2-n-),1.24(m,1h,ar-ch-(ch2)2),1.06(-o-c-(ch3)3),1.02,0.99(m,2h,ar-ch-(ch2)2).实施例7中间体3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑(8)的合成将tfa(2ml,26.9mmol)溶于无水dcm中,配制成20%的tfa溶液。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基-n-boc-哌啶)-5-环丙基异噁唑(400mg,0.83mmol)溶于dcm中,逐滴加入其中,室温下反应1h。饱和nahco3溶液终止反应,将反应液调成碱性,用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干,直接下投。实施例8中间体4-氨基苯甲酸甲酯的合成将对氨基苯甲酸(1g,7.3mmol)溶于ch3oh(10ml)中,冰浴下,缓慢滴加氯化亚砜(1ml,13.7mmol)。待氯化亚砜完全加入后,撤去冰浴,缓慢升温至50℃,反应2h。饱和nahco3溶液终止反应,将反应液调成碱性,用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。实施例93-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶苯基脲)-5-环丙基异噁唑(a1)的合成苯胺(29.2mg,0.31mmol)溶于无水dcm中。将三乙胺(0.1ml)和三光气(46.5mg,0.15mmol)在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑(120mg,0.31mmol)溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a1,产物:60mg,产率:40.5%。油状物。1hnmr(300mhz,chloroform-d)δ7.77–7.46(m,1h),7.46–7.38(m,1h),7.37–7.21(m,2h),7.09–6.98(m,0h),6.57(s,0h),3.61(ddd,j=11.9,7.1,3.9hz,1h),3.50(dt,j=7.6,3.7hz,0h),2.22–2.11(m,0h),1.76(td,j=7.9,3.6hz,1h),1.52(ddt,j=12.8,8.5,3.9hz,1h),1.37–1.22(m,1h),1.19–1.08(m,1h)。13cnmr(75mhz,cdcl3)δ171.9,159.5,155.0,146.9,139.1,131.8,131.2,128.8,127.0,123.1,123.1,120.8,120.0,119.3,111.6,77.5,77.1,76.7,73.5,59.0,41.4,30.5,8.1,7.6.hrms(esi)m/z[m+h]+502.2883,found502.1938。实施例103-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-4-甲氨基酰基苯基脲)-5-环丙基异噁唑(a2)的合成4-氨基-n-甲基苯甲酰胺溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a2,产率62.3%。油状物。1hnmr(dmso-d6,300mhz)δ8.86(s,1h,-conh-),7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.81(s,3h,-conhch3),2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2),13cnmr(75mhz,dmso-d6)δ172.05,158.23,155.39,154.84,147.00,139.18,131.66,131.04,128.40,126.75,122.77,120.85,119.81,111.45,77.88,77.18,76.26,74.65,73.60,59.20,41.61,30.50,8.25,7.36.hrms(esi)m/z[m+h]+559.2098,found559.2153。实施例113-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-3-甲氨基酰基苯基脲)-5-环丙基异噁唑(a3)的合成3-氨基-n-甲基苯甲酰胺溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a3,产率63.5%。油状物。1hnmr(dmso-d6,300mhz)δ8.86(s,1h,-conh-),7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.81(s,3h,-conhch3),2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,dmso-d6)δ172.1,159.3,157.4,154.8,147.0,139.2,131.7,131.0,128.4,126.8,122.8,120.8,119.8,111.4,77.8,77.5,76.3,74.6,73.6,59.2,41.6,30.5,8.0,7.3.hrms(esi)m/z[m+h]+559.2098,found559.2150。实施例123-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-3-甲氧基酰基苯基脲)-5-环丙基异噁唑(a4)的合成3-氨基苯甲酸甲酯溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a4,产率37.5%。油状物。1hnmr(dmso-d6,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.89(s,3h,-cooch3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,dmso-d6)δ172.1,159.3,159.2,154.8,148.0,139.2,131.7,131.0,128.4,126.8,122.8,120.8,119.8,111.4,77.7,77.1,76.3,73.6,59.2,41.6,30.5,8.8,7.9.hrms(esi)m/z[m+h]+532.1989,found532.2056。实施例133-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-3-乙氧基酰基苯基脲)-5-环丙基异噁唑(a5)的合成3-氨基苯甲酸乙酯溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a5,产率45.2%。油状物。1hnmr(dmso-d6,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),4.30(q,2h,-cooch2ch3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.30(t,3h,-cooch2ch3),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,cdcl3)δ172.1,159.3,158.4,154.8,147.0,139.2,131.6,131.1,128.4,126.8,122.8,120.8,119.8,111.4,77.8,77.1,76.3,73.6,59.2,41.6,30.5,8.0,7.3.hrms(esi)m/z[m+h]+573.2095,found573.2155。实施例143-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-4-甲氧基酰基苯基脲)-5-环丙基异噁唑(a6)的合成4-氨基苯甲酸甲酯溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a6,产率45.6%。油状物。1hnmr(dmso-d6,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.89(s,3h,-cooch3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,dmso-d6)δ172.05,159.31,158.63,154.84,147.00,139.16,131.66,131.05,128.40,126.75,122.77,120.83,119.81,111.41,77.78,77.08,76.26,73.60,59.20,41.61,30.50,7.95,7.33.hrms(esi)m/z[m+h]+573.2095,found573.2153.实施例153-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-4-乙氧基酰基苯基脲)-5-环丙基异噁唑(a7)的合成4-氨基苯甲酸乙酯溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a7,产率43.6%。油状物。1hnmr(dmso-d6,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),4.30(q,2h,-cooch2ch3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.30(t,3h,-cooch2ch3),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,dmso-d6)δ172.1,159.3,154.8,147.0,139.2,131.7,131.0,127.5,126.7,122.9,120.4,119.9,111.4,77.8,77.3,76.2,73.6,59.2,40.6,30.5,8.5,7.6.hrms(esi)m/z[m+h]+573.2095,found573.2153。实施例163-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-4-甲氧基苯基脲)-5-环丙基异噁唑(a8)的合成3-甲氧基苯胺溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a8,产率37.5%。油状物。1hnmr(cdcl3,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.61(s,3h,-och3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,cdcl3)δ172.1,159.7,154.8,152.0,149.2,131.7,131.0,128.4,126.8,122.8,120.4,119.9,111.9,77.8,77.1,76.3,73.6,59.2,41.6,30.5,8.0,7.3.hrms(esi)m/z[m+h]+532.1989,found532.2045。实施例173-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶-3-甲氧基苯基脲)-5-环丙基异噁唑(a9)的合成4-甲氧基苯胺溶于无水dcm中。将三乙胺和三光气在冰浴下溶于dcm后,逐滴加入,反应1h。减压除去多余的三光气。加入无水dcm溶解待用。将3-(2-三氟甲氧基苯基)-4-(4-氧甲基哌啶)-5-环丙基异噁唑溶于无水dcm中,逐滴加入,升温至35℃,反应过夜。反应液用ea和h2o萃取,饱和食盐水除水,无水na2so4干燥,旋干。柱层析纯化。得化合物a9,产率38.6%。油状物。1hnmr(cdcl3,300mhz)δ7.48-6.9(m,8h,ar),4.30(s,2h,ar-ch2-o-),3.61(s,3h,-och3),3.49(m,4h,-o-ch2-ch2-n-),3.37(m,1h,-o-ch-(ch2)2,2.22(m,1h,ar-ch-(ch2)2),1.68(m,4h,-o-ch2-ch2-n-),1.25(m,1h,ar-ch-(ch2)2),1.02,0.99(m,2h,ar-ch-(ch2)2)13cnmr(75mhz,cdcl3)δ172.1,159.3,154.8,147.0,139.2,131.7,131.0,128.4,126.8,122.8,120.8,119.8,116.4,78.0,77.4,76.3,73.6,58.2,41.6,30.5,8.6,7.9.hrms(esi)m/z[m+h]+532.1989,found532.2040。实施例181.mtt法测试3t3-l1细胞增殖实验将浓度约为1×105ml-1的3t3-l1细胞接种到96孔细胞培养板中,每孔100μl。在细胞培养箱中静置培养(37℃,5%co2)24h后,向每孔中加入100μl含不同浓度样品的培养基,使最终的样品浓度分别为5μmol/l、10μmol/l、20μmol/l和40μmol/l,每个浓度设置3个复孔。空白对照组为等体积含0.1%的dmso培养基。继续培养48h后,向每孔加20μl浓度为5mg/ml的mtt溶液,培养4h。4h后小心吸出孔中的液体,向每孔中加入150μldmso,于振荡器上低速振荡20min,用酶标检测仪检测各孔在490nm处吸光度od值,计算细胞抑制率。抑制率=[1-(实验组od值-空白组od值)/(对照组od值-空白组od值)]×100%3t3-l1前脂肪细胞的增殖活性实验结果见图1。可以看出大部分化合物在低浓度时(5μmol/l和10μmol/l)对细胞增殖有微弱抑制作用,个别化合物如a8等在5μmol/l浓度下具有促进细胞增殖的作用。在高浓度时(20μmol/l和40μmol/l),化合物对细胞都有一定的抑制活性,其中化合物a7在40μmol/l的浓度下,对细胞存活的抑制率达到50%以上。因此,我们选取化合物浓度为10μmol/l进行下一步的活性测试。2.对hepg2肝癌细胞内fxr和fxr下游基因mrna表达的影响选择处于指数生长期的hepg2肝癌细胞,用0.25%胰酶消化。消化完成后,用完全培养基终止。离心,加完全培养基使细胞重悬,在六孔板的每个孔中接种1×105个细胞。24h后,加入浓度为10μm的gw4064和目标化合物。待处理24h后,去除培养基,往六孔板中加pbs洗一遍细胞,去除pbs。加入1mltrizol,室温放置2min,将trizol转移至无rna酶的ep管中。加入氯仿200μl,用力振摇15s,室温静置15min,离心(4℃,12000g,15min)。离心后液体分三层,小心吸取上层无色液体,移入新的ep管中。加入等体积异丙醇,上下颠倒使之混匀,室温静置5min,离心(4℃,12000g,10min)。除去上清液,向沉淀中加入1ml75%乙醇,轻微振荡15s,离心(4℃,7500g,5min)。小心除去上清液,管内沉淀在超净台中鼓风静置干燥3-5min。加入50μldepc水溶解,nanodrop检测浓度,-80℃冰箱保存。将提取后的rna逆转录为cdna,逆转录实验步骤按照逆转录试剂盒说明书操作(takara,rr047)。逆转录所得cdna于steponeplus荧光定量pcr(abi)上反应。产物双链dna通过与sybrgreeni染料结合进行荧光定量,mrna的相对表达量通过公式2-δδct计算得到。实验结果表明,化合物a4能上调fxrmrna和shpmrna的转录水平,上调fxrmrna的转录水平比阳性药gw4064弱,上调shpmrna的转录水平与阳性药gw4064相当(图2-3),上调bsepmrna的转录水平比阳性药gw4064弱,下调bsrebp-1cmrna的转录水平比阳性药gw4064弱(图4-5)。3.fxr激动活性测试将对数生长期的hepg2细胞制备为细胞悬液(细胞浓度约为2×105个细胞/ml)接种于96孔板中,37℃、5%co2培养箱中培养。待细胞90%融合后,对于每孔细胞,取25μlopti-mem培养基稀释30ng质粒(gal4-fxrlbd)+50ng报告质粒pgl4.35+20ngrenilla荧光素酶对照载体+40ngpgemcarrierdna,轻轻混匀后于室温放置约5min;取25μlopti-mem培养基稀释0.35μl脂质体3000试剂,轻轻混匀后于室温放置约5min。混合两溶液,轻柔吹打均匀,室温静置约20min。将96孔板中的细胞用pbs冲洗细胞2遍,加入混合溶液轻轻混合均匀,细胞培养箱中继续培养约8h。8h后,将旧的培养基更换为含0.1%dmso或10μmol/l的gw4064和化合物a4dmem培养基,继续培养约18h后,检测荧光素酶活性时,操作间温度设定为22℃,避光。双荧光素酶报告基因系统检测步骤如下:裂解细胞,弃去旧的细胞培养液,每孔加100μl报告基因细胞裂解液,充分裂解细胞。细胞完成裂解后,每孔细胞转移至ep管,离心混合均匀。缓慢融解萤火虫荧光素酶检测试剂和海肾荧光素酶检测缓冲液至室温。海肾荧光素酶检测底物(100×)置于冰浴备用。按照1:100比例,向海肾荧光素酶检测缓冲液加入海肾荧光素酶检测底物(100×),配制成海肾荧光素酶检测工作液。开启glo-max20/20荧光检测仪,设置检测参数,检测间隔时间为2s,检测时间为10s。检测萤火虫荧光素酶活性值f(firelylueiferase)时,每个细胞样品加入100μl萤火虫荧光素酶检测试剂,混匀后将ep管立即放入bertholdlb941微孔板式多功能酶标仪,计算f值。加入100μl海肾荧光素酶检测工作液,混匀后立即放入bertholdlb941微孔板式多功能酶标仪,计算海肾荧光素酶活性值r(renillaluciferase)。完成所有样品测定后,由bertholdlb941微孔板式多功能酶标仪检测到的f值和r值,计算得出相对荧光素酶活性相对值,然后将各药物处理组的值对阴性对照孔的值进行归一化处理,拿得到的新的比值进行统计。实验结果表明,在10μmol/l的浓度下化合物a4能明显激活fxr(图6)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1