一种猪背膘厚的分子标记辅助选择方法及其应用
id no.1和seq id no.2所示的核苷酸序列。
12.本发明还提供了一种用于检测上述snp标记的试剂盒,其中,所述试剂盒包括引物对p1和p2,所述p1的核苷酸序列包括seq id no.1所示的核苷酸序列;
13.所述p2的核苷酸序列包括seq id no.2所示的核苷酸序列。
14.其中,所述试剂盒还包括pcr扩增体系,优选包括pcr缓冲液、dntp和dna聚合酶。
15.本发明还提供了一种猪背膘厚的分子标记辅助选择方法,其中,所述方法包括确定猪核心群中种猪位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记,并根据所述标记做出选择的步骤。
16.其中,所述确定猪核心群中种猪位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记的步骤包括对猪基因组dna进行扩增和测序的子步骤。
17.本发明所具有的有益效果包括:
18.(1)本发明所提供的与猪背膘厚相关的snp标记,可对背膘厚度进行早期选择,降低育种成本,缩短育种周期,促进猪的育种进程;
19.(2)本发明所提供的与猪背膘厚相关的snp标记,可以通过直接鉴定该标记来筛选更低背膘厚的猪品系,提高了养殖企业以及整个生猪养殖业的经济效益与社会价值;
20.(3)本发明提供的猪背膘厚的分子标记辅助选择方法,操作简单,准确性高,可以为猪生产性状的分子标记辅助选择提供科学依据。
附图说明
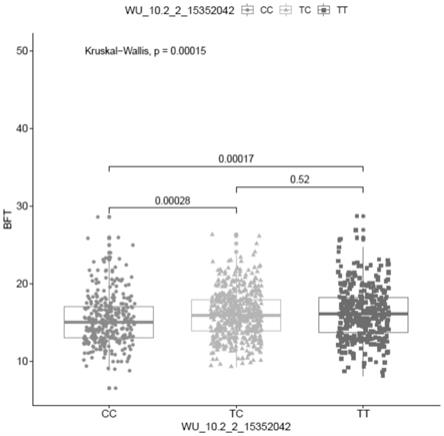
21.图1示出本发明实施例1中基因型效应的箱线图。
具体实施方式
22.下面通过优选实施方式和实施例对本发明进一步详细说明。通过这些说明,本发明的特点和优点将变得更为清楚明确。
23.在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。
24.全基因组关联分析是鉴定表型和基因型之间遗传联系的主要手段,可以用于发现影响背膘厚度的snps。
25.一方面,本发明提供了一种与猪背膘厚相关的snp标记,所述snp标记位于猪基因组2号染色体正义链上,其碱基类型为c或t。
26.其中,所述猪基因组2号染色体正义链核苷酸序列参照国际猪基因组10.2版本(sscrofa10.2)参考序列,所述snp标记位于2号染色体的15.35mb处,与qtl数据库比较显示,其处于平均背膘厚、瘦肉百分比、日采食量、心脏重量、第十肋骨背膘肉和肩部皮下脂肪厚度等性状相关的qtl区域。
27.优选地,所述与猪背膘厚相关的snp标记具体位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处。
28.其中,所述snp标记记为wu_10.2_2_15352042。
29.根据本发明一种优选的实施方式,所述位于国际猪基因组10.2版本参考序列2号
染色体第15,352,042个脱氧核苷酸处snp标记的cc基因型个体,其背膘厚度显著低于tc和tt基因型个体。
30.在本发明中,所述snp标记对应的基因型有三种:cc、tc和tt,cc是碱基c的纯合子,tt是碱基t的纯合子,tc为杂合子。
31.本发明人研究发现,猪该snp标记的cc基因型个体的背膘厚度显著低于tc基因型和tt基因型个体,从而能够根据该snp位点的基因型对猪的肉质性状进行遗传评估。由此,发明人确定,本发明所述的位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处的snp标记与猪背膘厚性状紧密相关,能够有效用于猪的分子标记辅助育种,具有早期筛选、节省时间、成本低廉、准确性高的优点。
32.其中,对snp位点进行基因分型可以在一定程度上排除表型选择中饲养环境、饲料、疾病等因素对猪优良基因的误淘和误选,增强目标性状选择准确性。
33.进一步地,本发明优选采用高密度snp芯片技术进行基因分型,其相对于传统的基因分型方法(如pcr、rflp等),可以在很短的时间周期内对大量的snp进行分型,效率高,其能极大地降低成本。
34.优选地,本发明中采用neogen公司的neogen_por80k芯片机芯基因分型。
35.本发明的又一方面,提供了一种用于检测上述snp标记的引物对,所述引物对包含seq id no.1和seq id no.2所示的核苷酸序列。
36.优选地,所述引物对为p1和p2,二者的核苷酸序列分别如seq id no.1和seq id no.2所示。
37.在本发明中,采用上述引物对能够有效对待测猪个体中含有上述与猪背膘厚相关的snp标记的核苷酸片段进行pcr扩增,进而通过测序实现对该snp位点的检测,确定该snp位点的基因型,进而确定待测猪个体的猪背膘厚度。
38.本发明的又一方面,提供了一种用于检测上述snp标记的试剂盒,所述试剂盒包括上述用于检测snp标记的引物对。
39.优选地,所述试剂盒还包括pcr扩增体系,优选包括pcr缓冲液、dntp和dna聚合酶。
40.本发明的又一方面,提供了一种上述snp标记、引物对或试剂盒在研究、检测、鉴定、调节、降低猪背膘厚或猪分子标记辅助育种方面的应用。
41.本发明的又一方面,提供一种上述snp标记的获取方法,所述方法包括以下步骤:
42.步骤1,选择猪群体,进行背膘厚测定。
43.在本发明中,优选在猪个体体重处于85~105kg范围时测定活体背膘厚(backfat thickness,bft),采用b超扫描测定群体中猪个体的倒数第3~4肋间处背膘厚,以毫米为单位。然后,采用河北省地方标准(db13/t 2065-2014)文件《种猪场场内生产性能测定技术规程》的遗传评估性状测定规程对采集的数据进行表型数据校正,具体按如下校正公式计算猪达100kg体重的活体背膘厚:
44.校正背膘厚=实测背膘厚
×
cf;
45.cf=a
÷
{a+[b
×
(实测体重-100)]}。
[0046]
其中,a、b为不同猪种背膘厚度校正系数,实测体重单位为kg。
[0047]
步骤2,提取猪个体的基因组dna,进行基因型分型。
[0048]
在本发明中,采用现有技术中常用的方法或试剂盒提取猪群体中每个个体的基因
组dna,优选采集猪耳组织进行基因组dna的提取。
[0049]
在提取基因组dna后,优选采用分光光度计和电泳对提取的猪基因组dna进行浓度测定和质量检测,其中,提取的dna的a260/a280比值在1.8~2.0,a260/a230比值在1.7~1.9判定为纯度合格;将浓度高于300ng/μl判定为浓度合格。
[0050]
优选地,将检测合格的dna利用高密度snp芯片进行基因型检测,如纽勤公司的neogen_por80k芯片,优选利用分型软件gencall version7.0.0进行。
[0051]
步骤3,对背膘厚数据和基因型分型数据进行质量控制。
[0052]
在本发明中,对背膘厚数据进行的质量控制为:清除表型值缺失的个体,清除表型值大于3倍标准差的个体。
[0053]
对基因型分型数据进行的质量控制为:清除基因型检出率小于95%的snp位点;清除检出率小于95%的个体;清除最小等位基因频率(maf)小于1%的个体;清除哈代-温伯格平衡(hwe)卡方检验p值小于1.0e-4的snp位点;清除性染色体上的snp位点。
[0054]
步骤4,进行背膘厚度全基因组关联分析。
[0055]
根据本发明一种优选的实施方式,优选采用r语言包gapit version 3对所有分型snp位点与校正背膘厚进行全基因组关联分析。
[0056]
其中,该软件包的统计模型为压缩混合线性模型,gapit的设计目的是在大数据集上准确地执行gwas和基因组预测,混合线性模型(mixed liner model,mlm)包括固定和随机效应,模型将种群结构作为固定效应,同时将个体纳入随机效应进行个体亲缘关系矩阵的构建,其统计分析模型如下:
[0057]
y=xβ+zu+e
[0058]
其中,y是观察到的表型的值;β是含有固定效应的未知值;u是来自个体或者系的多个背景qtl的随机加性遗传效应的未知值;x和z是已知的设计矩阵;e是未观察到的残差向量。
[0059]
通过上述关联分析,能够得到与猪背膘厚显著相关的snp位点,还需要进一步比较分析所得snp位点中不同基因型与校正背膘厚度的关联结果。
[0060]
在进一步优选的实施方式中,采用rstudio软件利用kruskal-wallis方法对所得snp位点的基因型数据和校正背膘厚数据进行差异显著性检验,以获得与猪校正背膘厚显著相关的基因型类型。
[0061]
本发明的又一方面,提供一种检测猪背膘厚度的方法,所述方法包括检测待测猪国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记的基因型的步骤。
[0062]
优选地,所述方法包括以下步骤:
[0063]
步骤i,提取待测猪的基因组dna。
[0064]
其中,采用现有技术中常用的方法或试剂盒提取待测猪的dna,优选采集猪耳组织。
[0065]
步骤ii,以基因组dna为模板,进行pcr扩增。
[0066]
其中,利用上述检测snp标记的引物对或包含该引物对的试剂盒进行pcr扩增,所得到的扩增产物包含位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处的snp标记。
[0067]
步骤iii,确定待测猪snp标记的基因型。
[0068]
其中,所述snp标记的检测方法不受特别限制,可以采用现有技术中常用的直接测序法、单链构象多态性聚合酶链式反应(pcr-sscp)、限制性片段长度多态性聚合酶链式反应(pcr-rflp)及飞行时间质谱等技术。其中,测序是一种准确性高、灵活性强、通量大、检测周期短的检测技术,可直接检测snp位点的基因型。因此,本发明中优选采用直接测序法进行检测,即将pcr扩增产物进行直接测序。
[0069]
其中,对测序的方法不做特别限制,只有能够获得pcr扩增产物的序列即snp标记所在的片段的核苷酸序列即可。例如可以采用hiseq2000、solid、454和单分子测序等方法,以便于高通量、快速、高效、准确地获得测序结果。
[0070]
基于测序结果,能够有效确定待测猪的所述snp标记的基因型。
[0071]
步骤iv,根据基因型确定待测猪的背膘厚度。
[0072]
其中,若待测猪位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记的基因型为cc,则其具有较低的背膘厚度;若待测猪位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记的基因型为tc或tt,则该待测猪具有较高的背膘厚。
[0073]
本发明所述的检测方法,能够快速、高效、准确地检测猪背膘厚,进而能够有效用于猪的分子标记辅助育种,从而能够早期实现短时间、低成本、高准确性地选育猪优良品种。
[0074]
本发明的又一方面,提供了一种猪背膘厚的分子标记辅助选择方法,所述方法包括确定猪核心群中种猪位于国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记,并根据所述标记做出选择的步骤。
[0075]
具体地,采用能够用于检测本发明的与猪背膘厚相关的snp标记的引物对或包含该引物对的试剂盒等,对种猪基因组dna进行扩增、测序,以确定种猪上述snp标记的基因型,进而继代选育国际猪基因组10.2版本参考序列2号染色体第15,352,042个脱氧核苷酸处snp标记的基因型为cc的个体,淘汰该snp位点基因型为tc或tt的个体,以逐代提高该位点的纯合基因型cc的频率,从而降低背膘厚。
[0076]
实施例
[0077]
以下通过具体实例进一步描述本发明,不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。
[0078]
实施例1
[0079]
1、试验猪群体
[0080]
本实施例所采用的试验猪群体来自于河北美神原种猪场的1177头猪,有大白猪、长白猪和杜洛克猪三个品种,其中,大白猪共597头,公猪2头,母猪595头,长白猪共379头,公猪15头,母猪364头,杜洛克猪共201头,公猪23头,母猪178头。
[0081]
2、背膘厚度的测定与校正
[0082]
在猪个体体重处于85~105kg范围时测定活体背膘厚(backfat thickness,bft),采用b超扫描测定群体中猪个体的倒数第3~4肋间处背膘厚,以毫米为单位。然后,采用河北省地方标准(db13/t 2065-2014)文件《种猪场场内生产性能测定技术规程》的遗传评估性状测定规程对采集的数据进行表型数据校正,具体按如下校正公式计算猪达100kg体重
的活体背膘厚:
[0083]
校正背膘厚=实测背膘厚
×
cf;
[0084]
cf=a
÷
{a+[b
×
(实测体重-100)]}。
[0085]
其中,a、b为不同猪种背膘厚度校正系数,实测体重单位为kg。
[0086]
三个猪品种的公母猪背膘厚度校正表如下表1所示:
[0087]
表1
[0088][0089]
3、提取猪基因组dna
[0090]
按照以下步骤进行:
[0091]
(1)收集猪耳组织,将其剪碎后放入一个1.5ml离心管后,加入200μl组织裂解液tl,用大口径枪头吹打混匀;
[0092]
(2)加入20μl的蛋白酶k(20mg/ml),剧烈颠倒轻摇充分混匀;
[0093]
(3)将裂解的耳组织放置在55℃水浴3小时或者直到组织消化完全,期间轻柔的振荡几次帮助裂解;
[0094]
(4)用一个1ml不带针头的一次性输液器吹打裂解物2-3次;
[0095]
(5)加入200μl结合液cb和100μl异丙醇,剧烈颠倒轻摇充分混匀;
[0096]
(6)13000rpm离心5分钟,将上清液加入一个吸附柱ac中,(吸附柱放入收集管中)10000rpm离心30秒,倒掉收集管中的废液;
[0097]
(7)加入500μl抑制物去除液ir,12000rpm离心30秒,弃废液;
[0098]
(8)加入700μl漂洗液wb(预先加入无水乙醇),12000rpm离心30秒,弃掉废液;
[0099]
(9)加入500μl漂洗液wb,12000rpm离心30秒,弃掉废液;
[0100]
(10)将吸附柱ac放回空收集管中,13000rpm离心2分钟,尽量除去漂洗液,以免漂洗液中残留乙醇抑制下游反应;
[0101]
(11)取出吸附柱ac,放入一个干净的离心管中,在吸附膜的中间部位加100μl洗脱缓冲液eb(洗脱缓冲液事先在65-70℃水浴中预热),室温放置3-5分钟,12000rpm离心1分钟;将得到的溶液重新加入离心吸附柱中,室温放置2分钟,12000rpm离心1分钟;洗脱体积越大,洗脱效率越高,如果需要dna浓度较高,可以适当减少洗脱体积,但是最小体积不应少于50μl,体积过小降低dna洗脱效率,减少dna产量;
[0102]
(12)经过nanodrop-100分光光度计检测质量与浓度后将浓度统一稀释到50ng/μl,可以暂存于2-8℃,如果要长时间存放,可以放置在-20℃,为dna分型做准备。
[0103]
4、基因分型
[0104]
取检测合格的dna,利用纽勤公司的neogen_por80k芯片(商品目录号:902148)检测每个个体的基因型。
[0105]
5、数据的质量控制
[0106]
在背膘厚表型值测定过程中,清除表型值缺失的个体,清除表型值大于3倍标准差的个体;
[0107]
基因分型过程中,清除基因型检出率小于95%的snp位点;清除检出率小于95%的个体;清除最小等位基因频率小于1%的个体;清除哈代-温伯格平卡方检验p值小于1.0e-4的snp位点;清除性染色体上的snp位点。
[0108]
6、统计snp位点的基因型在试验猪群体中的分布,结果如表2所示。
[0109]
表2
[0110][0111]
由表2可知,tc基因型为实验群体的优势基因型。
[0112]
7、采用r语言包gapit version 3对所有分型snp位点与校正猪背膘厚进行全基因组关联分析,采用的统计分析模型如下:
[0113]
y=xβ+zu+e
[0114]
其中,y是观察到的表型的值;β是含有固定效应的未知值;u是来自个体或者系的多个背景qtl的随机加性遗传效应的未知值;x和z是已知的设计矩阵;e是未观察到的残差向量。
[0115]
统计结果表明,位点wu_10.2_2_15352042与猪背膘厚性状显著相关。
[0116]
8、采用rstudio软件对基因型数据和表型数据利用kruskal-wallis方法进行差异显著性检验,其中,fdr_adjusted_p-values,即校正后的p值,0.1<p-value<0.05表明差异显著。关联分析结果如表3所示:
[0117]
表3
[0118][0119]
从表3可以看出,cc基因型猪的背膘厚度显著低于tc和tt基因型个体。
[0120]
进一步地,采用r软件绘制三种基因型效应的箱线图,结果如图1所示。
[0121]
由图1可知,cc基因型的个体校正背膘厚比tc基因型的低0.69mm,比tt基因型的低0.86mm。
[0122]
综上所述,wu_10.2_2_15352042位点cc纯合型个体在背膘厚度方面具有优势。wu_10.2_2_15352042位点可以作为背膘厚度的遗传标记,应用于背膘厚度性状的分子标记辅助选择,提高猪的背膘厚度选择的准确性。
[0123]
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1