一种7β-甲氧基头孢美唑的分离纯化方法与流程
一种7
β-甲氧基头孢美唑的分离纯化方法
技术领域
1.本发明属于医药技术领域,具体涉及一种7β-甲氧基头孢美唑的分离纯化方法。
背景技术:
2.头孢美唑,化学名为(6r,7s)-7-[(氰甲基硫基)乙酰氨基]-7-甲氧基-3-[1-甲基-1h-四氮唑-5-硫甲基]-8-氧化-5-硫杂-1-氮杂环[4.2.0]辛-2-烯-2-羧酸,是第二代半合成头霉素类抗菌药物,具有广谱、高效、低毒的作用,其化学结构式见图1。
[0003]
头孢美唑的作用机制与头孢菌素类药相似,主要是通过干扰增殖期细菌细胞壁的合成而发挥杀菌作用。头孢美唑的临床应用十分广泛,但是由于该化合物结构复杂,其工艺步骤多,合成难度大,部分中间体稳定性差,容易产生杂质,杂质分离鉴别难度更大。
[0004]
新药研发必须遵守“安全、有效、质量可控”的基本原则。各国或组织就杂质的限度论证先后出台了多项指导原则,核心是要求将药物中实际存在的以及潜在的杂质能够被很好的控制,并尽可能将其去除,从而使得我们的药物更安全。因此,杂质的制备分离对于药物质量的控制及人们的用药安全都有着十分深远的意义。
[0005]
先前kunio atsumi等人(tetrahedron letters, 23, 2977-2980)发现在大量头孢美唑中间体合成的过程中不可避免的会产生微量的头孢美唑7β-甲氧基异构体。随着本技术人多年来对头孢美唑生产制备工艺及产品质量研究的深入,改变原有合成路线后,粗品经hplc-ms检测,确定含有头孢美唑的一个同分异构体,通过大量工作成功将其分离纯化,并经1h-nmr、
13
c-nmr鉴定,确定为头孢美唑7β-甲氧基异构体,其化学结构式见图2。目前,得到该异构体对于头孢美唑的药品质量研究和质量控制有重要意义。目前也未见其他文献中有关7β-甲氧基头孢美唑该杂质的分离纯化方法。
技术实现要素:
[0006]
本发明的目的在于提供一种头孢美唑新型杂质7β-甲氧基头孢美唑的分离纯化方法,利用制备液相对该新型杂质进行分离纯化。
[0007]
本发明的目的是通过下列技术方案实现的:一种7β-甲氧基头孢美唑的分离纯化方法,该分离纯化方法经过二次分离纯化,具体步骤如下:1)样品溶解:将含有7β-甲氧基头孢美唑异构体混合物溶解在乙腈溶剂中,得到混合溶液,并控制该混合溶液的浓度,过滤后注入反相高效制备液相色谱;2)第一次分离纯化:色谱柱选用十八烷基硅烷键合硅胶填料,流动相为tfa水溶液和乙腈,当流动相平衡系统后,进行等度洗脱,分段收集目标组分;3)第二次分离纯化:色谱柱选用十八烷基硅烷键合硅胶填料,流动相为tfa水溶液和乙腈,先用乙腈平衡柱子,然后将第一次分离纯化后的目标组分上样到分离柱上,再进行等度洗脱,分段收集目标峰流分;4)浓缩和冻干:将第二次分离纯化收集到的纯度合格的目标组分7β-甲氧基头孢美唑
低温减压蒸馏,除去大部分有机溶剂后,放入冻干机,进行冷冻干燥,即得到目标产物7β-甲氧基头孢美唑。
[0008]
优选的,步骤1)中所述的含有7β-甲氧基头孢美唑异构体的混合物与乙腈溶剂的质量体积比为(0.5g~1 g):10ml,所述混合溶液的浓度为5~100 mg/ml。
[0009]
优选的,步骤2)和步骤3)中,所述的色谱柱尺寸为20
×
250 mm,所述的十八烷基硅烷键合硅胶填料粒度为10 μm。
[0010]
更优选的,步骤2)中,流动相为0.1% tfa:乙腈=75:25,流速 20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm。
[0011]
更优选的,步骤3)中,流动相为0.1% tfa:乙腈=68:32,流速 20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm,先用乙腈占比为10%的流动相平衡柱子10 min。
[0012]
本发明的技术优点在于:结构相似的同分异构体之间的拆分大多都是不易完成的,而本发明方法成功地从头孢美唑异构体混合物中分离得到7β-甲氧基头孢美唑,纯度高达98%,重现性好,便于操作。该杂质对于头孢美唑的质量研究具有重要意义,为其杂质研究提供了优质的对照品,以便于更好的控制药物质量。
附图说明
[0013]
图1为头孢美唑化学结构式。
[0014]
图2为7β-甲氧基头孢美唑化学结构式。
[0015]
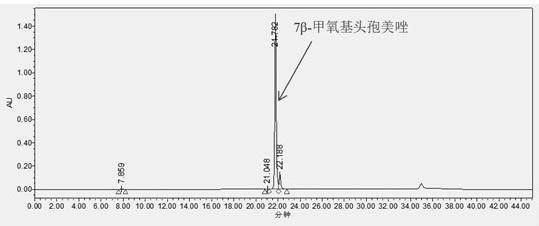
图3为对比例1分段收集的制备组分的hplc分析图谱。
[0016]
图4为对比例2分段收集的制备组分的hplc分析图谱。
[0017]
图5为实施例3中第一次分离纯化分段收集的制备组分的hplc分析图谱。
[0018]
图6为实施例3中第二次分离纯化分段收集的制备组分的hplc分析图谱。
[0019]
图7为实施例3得到的目标杂质7β-甲氧基头孢美唑1h-nmr谱图。图8为实施例3得到的目标杂质7β-甲氧基头孢美唑
13
c-nmr谱图。
具体实施方式
[0020]
下面结合具体实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
[0021]
本发明一种7β-甲氧基头孢美唑的分离纯化方法,采用反向高效制备液相色谱法对头孢美唑异构体混合物进行分离纯化;所述的反相高效制备液相色谱法条件为:制备液相柱为c18液相柱,检测器为紫外检测器,检测波长为190~400nm,流速为1~30 ml/min,温度为室温,第一次分离流动相为0.1%tfa
ꢀ-
乙腈,乙腈占比20%~30%。
[0022]
所述的头孢美唑异构体混合物用乙腈溶解,浓度为1~100 mg/ml,过滤,然后注入反相高效制备液相中进行分离纯化。
[0023]
由于第一次分离后主峰后杂质较大需进行第二次纯化分离,制备液相柱为c18液相柱,检测器为紫外检测器,检测波长为190~400 nm,流速为1~30 ml/min,温度为室温,压力为4 mpa,第二次分离流动相为0.1%tfa
ꢀ-
乙腈,乙腈占比30%~35%。第二次分离纯化
时,先用10%乙腈平衡柱子10 min,然后将浓缩后的制备组分用泵上样到分离柱上,再进行洗脱,分段收集目标组分。
[0024]
本发明反相高效制备液相色谱法分离出的制备组分通过低温减压蒸馏、真空冻干机冷冻干燥获得精制纯化物。
[0025]
本发明优选的制备液相色谱柱为ht-ods-p (10 μm,20x 250 mm)。
[0026]
实施例1对含有7β-甲氧基头孢美唑异构体混合物分离纯化,分离纯化方法包括如下步骤:1、样品溶解:将1 g头孢美唑异构体混合物溶解在10 ml乙腈溶剂中,得到混合溶液,混合溶液的质量浓度为100 mg/ml,过滤后注入反相高效制备液相色谱。
[0027]
2、第一次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=80:20,流速 20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm,当流动相平衡系统后,进行等度洗脱,分段收集目标组分。
[0028]
3、纯度检测:将分段收集的制备组分进行hplc纯度检测。
[0029]
4、第二次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=70:30,流速 20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm。先用10%的乙腈平衡柱子10 min,然后将第一次纯化分离组分浓缩后,上样到分离柱,再进行等度洗脱,分段收集目标峰。
[0030]
5、纯度检测:将分段收集的制备组分进行hplc纯度检测,7β-甲氧基头孢美唑纯度为93%。
[0031]
6、浓缩、冻干:将收集到的目标组分7β-甲氧基头孢美唑低温减压蒸馏除去大部分有机溶剂后,放入真空冻干机,进行冷冻干燥,即可得到目标杂质7β-甲氧基头孢美唑。
[0032]
实施例2对含有7β-甲氧基头孢美唑异构体混合物分离纯化,分离纯化方法包括如下步骤:1、样品溶解:将1 g头孢美唑异构体混合物溶解在10 ml乙腈溶剂中,得到混合溶液,混合溶液的质量浓度为100 mg/ml,过滤后注入反相高效制备液相色谱。
[0033]
2、第一次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=70:30,流速20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm,当流动相平衡系统后,进行等度洗脱,分段收集目标组分。
[0034]
3、纯度检测:将分段收集的制备组分进行hplc纯度检测。
[0035]
4、第二次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=65:35,流速 20 ml/min,进样量 2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm。先用10%的乙腈平衡柱子10 min,然后将第一次纯化分离组分浓缩后,上样到分离柱,再进行等度洗脱,分段收集目标峰。
[0036]
5、纯度检测:将分段收集的制备组分进行hplc纯度检测,7β-甲氧基头孢美唑纯度为95 %。
[0037]
6、浓缩、冻干:将收集到的目标组分7β-甲氧基头孢美唑低温减压蒸馏除去大部分有机溶剂后,放入真空冻干机,进行冷冻干燥,即可得到目标杂质7β-甲氧基头孢美唑。
[0038]
实施例3对含有7β-甲氧基头孢美唑异构体混合物分离纯化,分离纯化方法包括如下步骤:1、样品溶解:将1 g头孢美唑异构体混合物溶解在10 ml乙腈溶剂中,得到混合溶液,混合溶液的质量浓度为100 mg/ml,过滤后注入反相高效制备液相色谱。
[0039]
2、第一次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=75:25,流速 20 ml/min,进样量 2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm,当流动相平衡系统后,进行等度洗脱,分段收集目标组分。
[0040]
3、纯度检测:将分段收集的制备组分进行hplc纯度检测,如图5所示。
[0041]
4、第二次分离纯化:色谱柱尺寸为20
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度10 μm,流动相为0.1% tfa:乙腈=68:32,流速 20 ml/min,进样量2 ml,温度为室温,压力为4 mpa,采用紫外检测器,检测波长为220 nm。先用10%的乙腈平衡柱子10 min,然后将第一次纯化分离组分浓缩后,上样到分离柱,再进行等度洗脱,分段收集目标峰。
[0042]
5、纯度检测:将分段收集的制备组分进行hplc纯度检测,如图6所示。由图6可知,目标组分7β-甲氧基头孢美唑纯度达到98%。
[0043]
6、浓缩、冻干:将收集到的纯度合格的目标组分7β-甲氧基头孢美唑低温减压蒸馏除去大部分有机溶剂后,放入真空冻干机,进行冷冻干燥,即可得到目标杂质7β-甲氧基头孢美唑。
[0044]
7、结构确证:经1h-nmr、13c-nmr确证,如图7、8所示。
[0045]
1h-nmr(400mhz, dmso):δ9.92 (s, 1h,
ꢀ-
oh), 5.13 (s, 1h,
ꢀ-
ch-), 4.35-4.20 (dd, 2h,
ꢀ-
ch
2-), 3.93(s, 3h,
ꢀ-
ch3), 4.35-4.21 (dd, 2h,
ꢀ-
ch
2-), 3.78 (s, 2h,
ꢀ-
ch
2-), 3.76-3.47 (dd, 2h,
ꢀ-
ch
2-), 3.38(s, 3h,
ꢀ-
ch3)。
[0046]
13c-nmr(400mhz, dmso):δ169.5, 163.0, 160.4, 153.4, 126.6, 126.1, 118.2, 92.7, 63.5, 52.9, 35.9, 34.8, 34.2, 27.4, 17.2。
[0047]
esi-ms(m/z):494.04[m+na]+。
[0048]
申请人尝试进行一次分离纯化,如对比例1和2,但一次分离后主峰后杂质较大,分离纯化效果较差。
[0049]
对比例1头孢美唑异构体混合物的分离纯化,分离纯化方法包括如下步骤:1、样品溶解:将50 mg 头孢美唑异构体混合物溶解在10 ml 0.1% tfa:乙腈= 65:35的流动相中,质量浓度为5 mg/ml,过滤后注入反相高效制备液相色谱。
[0050]
2、分离纯化:色谱柱尺寸为4.6
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度5μm,流动相为0.1% tfa:乙腈= 65:35,流速为1 ml/min,进样量5 μl,温度为室温,压力为2200 psi,采用紫外检测器,检测波长为254 nm,当流动相平衡系统后,进行等度洗脱,分段收集目标组分。
[0051]
3、纯度检测:将分段收集的制备组分进行hplc纯度检测,发现未能有效分离目标
组分7β-甲氧基头孢美唑,如图3所示。
[0052]
对比例2头孢美唑异构体混合物的分离纯化,分离纯化方法包括如下步骤:1、样品溶解:将50 mg 头孢美唑异构体混合物溶解在10 ml 0.1% tfa:乙腈= 70:30的流动相中,质量浓度为5 mg/ml,过滤后注入反相高效制备液相色谱。
[0053]
2、分离纯化:色谱柱尺寸为4.6
×
250 mm,柱内装有十八烷基硅烷键合硅胶填料,填料粒度50 μm,流动相为0.1% tfa:乙腈=70:30,流速 1 ml/min,进样量5 μl,温度为室温,压力为2200 psi,采用紫外检测器,检测波长为254 nm,当流动相平衡系统后,进行等度洗脱,分段收集目标组分。
[0054]
3、纯度检测:将分段收集的制备组分进行hplc纯度检测,发现未能有效分离目标组分7β-甲氧基头孢美唑,如图4所示。
[0055]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1