一种多肽类衍生物及其在医药领域的用途的制作方法
本发明涉及医药领域,具体涉及一种多肽类衍生物及其在医药领域的用途。
背景技术:
镇痛药主要作用于中枢或外周神经系统,选择性抑制和缓解各种疼痛,减轻疼痛而致恐惧紧张和不安情绪疼痛的药物。从产品类型来看,目前全球镇痛市场以阿片类药物和非甾体抗炎药(non-甾体抗炎药,nsaids)为主,2015年共占市场总收入的52%以上。据根据美国透明市场研究(tmr)的一份报告预测,到2025年底全球镇痛市场将达到882.534亿美元。
传统μ阿片样物质受体激动剂(如吗啡及其衍生物)是临床解除剧烈疼痛的主要药物,全世界使用量最大的强效镇痛剂,是治疗慢性关节炎、炎症性神经痛、术后疼痛以及各种癌症引起的中到重度疼痛的最有效的药物。但全身施用传统阿片类镇痛药物会产生副作用,如呼吸抑制、药物成瘾、便秘、恶心、意识模糊和产生耐受等。哌啶类(哌替啶,芬太尼类)也是μ阿片样物质受体激动剂,药理作用与吗啡相同,临床应用与吗啡也相同。但哌替啶镇静、麻醉作用较小,呼吸抑制作用较吗啡弱,不良反应比吗啡小。其他常见的μ阿片样物质受体激动剂包括氨基酮类(美沙酮,右丙氧芬)、环己烷类衍生物(曲马朵)、氨基四氢萘类(地佐辛)。目前还有很多处于临床前和临床阶段μ阿片样物质受体激动剂。
κ阿片样物质受体(kor)由380个氨基酸组成,强啡肽是其内源性配体。其在感觉神经元、背根神经节细胞和初级传入神经元末梢中均有表达,与痛觉、神经内分泌、情感行为和认知等主要的生理活动。κ阿片样物质受体激动剂与μ阿片样物质受体激动剂不同,其不会导致呼吸抑制和便秘,并且研究表明其成瘾性更低。在机体正常情况下外周施用阿片样物质受体激动剂没有任何镇痛效果,在有炎症或组织损伤时,外周阿片样物质受体功能增强,在施用阿片样物质受体激动剂后发挥镇痛效果。此外,机体对于κ阿片样物质受体激动剂也不容易产生耐受。
专利(wo2013184794)报道了一种的全新的多肽类κ阿片样物质受体激动剂cr845。该分子结构中含有d构型氨基酸的四肽,在临床试验中都显示了很强的长效镇痛活性和较小的成瘾性。cr845的适应症比较广,已经启动的临床适应症包括:急性疼痛,尿毒症瘙痒,腹部手术后疼痛,骨关节炎,髋部、肌肉骨骼疾病,风湿性疾病,术后疼痛,瘙痒症,慢性肾病等。然而cr845临床iii期试验显示其会引起高血钠症等一些副作用,因此获得活性更好、副作用更小、成药性更好的新型κ阿片样物质受体激动剂仍然具有吸引力。值得注意的是,目前全球尚无外周选择性κ受体激动剂类镇痛药获批上市。因此一旦副作用更小的κ阿片样物质受体(kor)激动剂获批上市,必定成为具有很强竞争力的镇痛抗炎产品,将对现有市场产生极大的冲击。
技术实现要素:
为了解决上述技术问题,本发明公开一种多肽类衍生物,该多肽类衍生物作为κ阿片样物质受体激动剂,镇痛活性更好、副作用更少,本发明还公开该多肽类衍生物在医药领域的用途。
本发明通过下述技术方案实现:
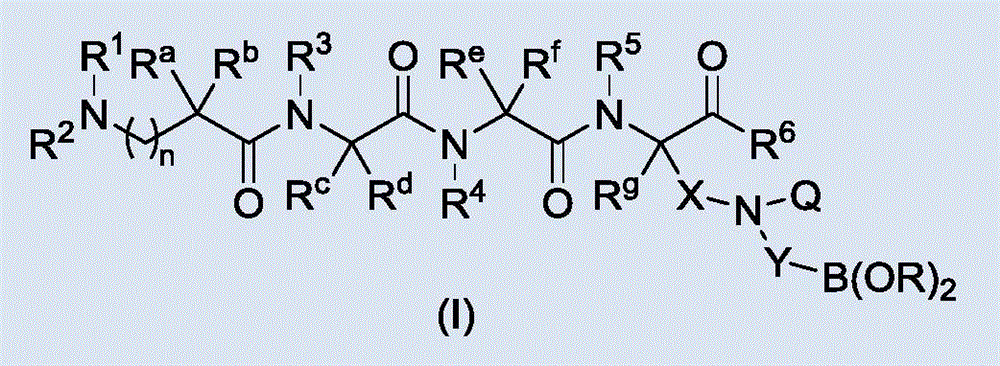
一种多肽类衍生物,其结构如通式(i)所示:
其中,所述通式(i)中,n为选自0到6中的任意整数;
r1选自氢原子、烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、邻苯二甲酰基、对甲苯磺酰基、邻硝基苯磺酰基、对硝基苯磺酰基、叔丁氧羰基、苄氧羰基、9-芴甲氧羰基、烯丙氧羰基、三甲基硅乙基氧羰基、c1~c8烷氧羰基、c1~c8酰基、三氟乙酰基、芳基甲酰基、三苯甲基、苄基,2,4-二甲氧基苄基和对甲氧基苄基,其中,所述的烷基、烷氧基、卤代烷基、环烷基、环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基可被选自烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基、炔基、羟基、氨基、硝基、氰基、羧基、酯基、酰胺基、巯基中的一个或多个取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s、b、p的杂原子;
r2、r3、r4、r5、ra、rc、re、rg各自独立地选自氢原子和c1~c10烷基;
rb、rd、rf各自独立地选自下列取代基:
氢原子、烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基、炔基、羟基、氨基、硝基、氰基、羧基、酯基、酰胺基,其中所述的烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基、炔基可被选自烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基、炔基、羟基、氨基、硝基、氰基、羧基、酯基、酰胺基、巯基中的一个或多个取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s、b、p的杂原子;
r6选自以下基团:
进一步的,所述通式(i)中,r1连同r2、ra和rb中的一个或多个原子成环,包括但不限于以下基团:
进一步的,所述通式(i)中,q=h,或者q=z-b(or)2;
x、y、z选自下列取代基:
c1~c12烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基烷基、炔基烷基,其中上述取代基可被c1~c12烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基烷基、炔基烷基、酯基、酰胺基、卤素、硝基、氰基、巯基、羟基、烷氧基、氨基、烯基、炔基中的一个或多个取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s、p的杂原子。
进一步的,x-n、z-n包含但不仅限于以下基团:
其中,n1、n2选自0到10中的任意整数;
b(or)2为以下取代基中的任一一种,且两个r基团中的部分原子能够连接成环状取代基,如下所示:
进一步的,r7选自氢原子、烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、-or7a、-c(o)r7a和-c(o)or7a,其中所述的烷基、烷氧基、卤代烷基、环烷基、环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基可被选自烷基、烷氧基、卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、烯基、炔基、羟基、氨基、硝基、氰基、羧基、酯基、酰胺基、巯基中的一个或多个取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s、b、p的杂原子;
r7a选自氢原子、烷基、氨基、烷氧基、羟基烷基卤代烷基、环烷基,环烷基烷基、杂环烷基、杂环烷基烷基、芳基、芳基烷基,其中所述的烷基、环烷基、杂环烷基、芳基、芳基烷基、杂芳基可被选自烷基、卤素、羟基、氨基、硝基、氰基、烷氧基、环烷基,羟烷基、杂环烷基、芳基、杂芳基中的一个或多个取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s的杂原子;
r8、r9选自f、cf3、c1~c烷基、c2~c6烯基、c2~c6炔基或3~8元杂环烷基,所述烷基、烯基、炔基或杂环烷基可被0-5个选自f、cl、br、i、oh、cf3、硝基、氰基、c1~c6烷基、c1~c6烷氧、c2~c6烯基、c2~c6炔基、c3~c8碳环基或3~8元杂环烷基的取代基所取代,所述的杂环烷基或杂芳基含有1到4个选自n、o、s的杂原子;
r10选自氢原子、氨基、取代氨基、氰基、羧基、-coor’、-conr’r”、-co(ch2)m-or’、-or’、-(ch2)mor’、-so2nr’r”、-(ch2)mcoor’、-(ch2)mconr’r”、-(ch2)mnr’r”、-nh(ch2)mcoor’、-(ch2)mconhr、-co(ch2)m-nr’r”、-co(ch2)m-coor’、-co(ch2)m-conr’r”、-co(ch2)m-conhnr’r”、-(ch2)m-conhnr’r”、-(ch2)m-nhnr’r”、-(ch2)mcn、-chr’coor’、-cr”r’coor”’、烷基芳基、芳基、c1~c10烷基、脒基、c1~c6烷基取代脒基,其中,r’、r”和r”’可被选自氢原子、c1~c10烷基、支链烷基、烷基芳基、芳基中的一个或多个取代基所取代,或者r’、r”组合成4~6元环;
m,m1,m2,m3,m4为选自0到6中的任意整数。
本发明设计并合成得到了一系列含有氨基硼酸结构片段的新型多肽类衍生物,其中,含硼化合物具有微妙的特性,能够可逆的与蛋白质靶标作用,硼酸基团与多肽结合,得到的多肽类衍生物作为κ阿片样物质受体激动剂药物,镇痛活性更好,而且由于硼酸基团具有独特的大极性和水溶性特征,脑通透性会更低,因此副作用更少。
优选的,一种多肽类衍生物,其结构如通式(ii)所示:
优选的,所述通式(ii)包括但不限于以下结构的化合物:
本发明所述通式(i)、(ii)的化合物的制备方法选自但不限于以下方法:
步骤一:化合物sm1(2-氯三苯甲基氯树脂)与化合物sm2(boc-4-氨基-1-fmoc-哌啶-4-羧酸)通过缩合反应得到化合物sm3;
步骤二:化合物sm3通过选择性脱fmoc保护基后,与fmoc-lys(alloc)-oh通过缩合反应得到化合物sm4;
步骤三:化合物sm4通过选择性脱fmoc保护基后,与fmoc-val-oh通过缩合反应得到化合物sm5;
步骤四:化合物sm5通过选择性脱fmoc保护基后,与fmoc-phe-oh通过缩合反应得到化合物sm6;
步骤五:化合物sm6通过选择性脱fmoc保护基后,与fmoc-phe-oh通过缩合反应得到化合物sm7;
步骤六:化合物sm7通过选择性脱alloc保护基后得到化合物sm8;
步骤七:化合物sm8通过在三氟乙醇条件下裂解得到化合物sm9;
步骤八:化合物sm9与sm11通过还原胺化反应得到化合物sm10;
步骤九:化合物sm10通过选择性脱fmoc保护基后得到化合物sm12;
步骤十:化合物sm10通过酸性条件下脱boc保护基后得到目标化合物tm。
一种多肽类衍生物在医药领域的用途。
本发明的多肽类化合物包括多肽类化合物的立体异构体、多晶型物、溶剂合物、代谢物、前药或药学上可接受的盐或酯、或其硼酸聚合物,以及一种或者多种以上的药学上可接受的载体和/或赋形剂。
本发明所使用的物料缩写含义如下表:
本发明与现有技术相比,具有如下的优点和有益效果:
本发明一种多肽类衍生物,具体为一系列含有氨基硼酸结构片段的新型多肽类衍生物,其中,含硼化合物具有微妙的特性,能够可逆的与蛋白质靶标作用,硼酸基团与多肽结合,得到的多肽类衍生物作为κ阿片样物质受体激动剂药物,镇痛活性更好、副作用更少。
附图说明
此处所说明的附图用来提供对本发明实施例的进一步理解,构成本申请的一部分,并不构成对本发明实施例的限定。在附图中:
图1为本发明多肽类衍生物通式(i);
图2为本发明实施例1工艺流程图;
图3为本发明实施例2工艺流程图;
图4为本发明实施例3工艺流程图;
图5为本发明实施例4工艺流程图;
图6为本发明实施例5工艺流程图;
图7为本发明实施例6工艺流程图;
图8为本发明实施例7工艺流程图;
图9为本发明实施例8工艺流程图;
图10为本发明实施例9工艺流程图;
图11为本发明实施例10工艺流程图;
图12为本发明实施例12工艺流程图;
图13为本发明实施例13工艺流程图;
图14为本发明实施例14工艺流程图;
图15为本发明实施例15工艺流程图;
图16为本发明实施例16工艺流程图;
图17为本发明实施例19工艺流程图;
图18为本发明实施例20工艺流程图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例和附图,对本发明作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本发明的限定。
实施例1
如图2所示,tm1可通过以下工艺步骤制得:
步骤一:合成中间体sm3
在室温条件下用dcm(20ml)溶胀2-ctcresin(取代度为0.993mmol/g,2g),溶胀时间15min,抽掉溶剂,将sm2(1.120g,2.4mmol)和diea(0.516g,4.0mmol)的dcm(15ml)混合溶液加入溶胀的树脂中,室温条件下反应2h;再加入甲醇(2ml)和diea(1ml),继续反应0.5h;抽干溶剂,再用dcm(30ml)洗三次,最后用dmf(30ml)洗三次,直接将树脂往下一步投反应。
步骤二:合成中间体sm4
向步骤一中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-lys(boc)-oh(1.809g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应。
步骤三:合成中间体sm5
向步骤二中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-leu-oh(1.421g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应。
步骤四:合成中间体sm6
向步骤三中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-phe-oh(1.551g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应。
步骤五:合成中间体sm7
向步骤四中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-phe-oh(1.547g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应。
步骤六:合成中间体sm8
向步骤五中树脂加入pd(pph3)4(200mg)和苯硅烷(0.435g,4.0mmol)的dcm(20ml),在室温条件下反应3h,抽干溶剂,并用dcm(30ml)洗涤6次,最后用甲醇(30ml)收缩3次,将树脂烘干得到4.2g中间体sm8;抽干直接用于下一步反应。
步骤七:合成中间体sm9
在室温条件下将sm8(4.2g)加入三氟乙醇/dcm(50ml,v=/v=1/4)在中,在室温条件下反应2h,抽滤,并用dcm(30ml)洗2次,将有机相浓缩到5ml左右;将浓缩液滴加到100ml的甲基叔丁基醚中搅拌沉降得到2.1g产品,经lcms(ms(m+h+)=1002.5检测为目标产品。
步骤八:合成中间体sm10-1
在室温条件下,将乙酸(120mg,1.0mmol),sm9(500mg,0.5mmol)和sm11-1(71mg,0.5mmol)溶液1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(212mg,1.0mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm10-1(322mg)。
esi-ms(m/z):1074.6(m+h+)
步骤九:合成中间体sm12-1
在室温条件下,将sm10-1(250mg,0.25mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到的粗品经过制备hplc纯化得到中间体sm12(188mg)。
esi-ms(m/z):852.5(m+h+)
步骤十:合成产品tm1
在室温条件下,将sm12-1(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到的粗品经过制备hplc纯化后,冻干得到产品tm1(32mg)。
esi-ms(m/z):752.4(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.36-7.12(m,10h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.30-2.88(m,12h),2.45(m,2h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h),0.69(m,2h)。
实施例2
如图3所示,tm2可通过以下工艺步骤制得:
步骤一:合成中间体sm11-2
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-2(46mg,0.3mmol)溶于1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm11-2(211mg)。
esi-ms(m/z):1136.6(m+h+)
步骤二:合成中间体sm12-2
在室温条件下,将sm11-2(190mg,0.18mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到的粗品经过制备hplc纯化得到中间体sm12-2(136mg)。
esi-ms(m/z):914.5(m+h+)
步骤三:合成产品tm2
在室温条件下,将sm12-2(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品tm2(53mg)。
esi-ms(m/z):814.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.82(s,2h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h)。
实施例3
如图4所示,tm3可通过以下工艺步骤制得:
步骤一:合成中间体sm11-3
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-3(45mg,0.3mmol)溶于1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm11-3(205mg)。
esi-ms(m/z):1136.6(m+h+)
步骤二:合成中间体sm12-3
在室温条件下,将sm11-3(190mg,0.18mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经过制备hplc纯化得到中间体sm12-3(142mg)。
esi-ms(m/z):914.5(m+h+)
步骤三:合成产品tm3
在室温条件下,将sm12-3(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品tm3(37mg)。
esi-ms(m/z):814.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.92(s,2h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h)。
实施例4
如图5所示,tm4可通过以下工艺步骤制得:
步骤一:合成中间体sm11-4
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-4(45mg,0.3mmol)溶于1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm11-4(160mg)。
esi-ms(m/z):1136.6(m+h+)
步骤二:合成中间体sm12-4
在室温条件下,将sm11-4(160mg,0.15mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经过制备hplc纯化得到中间体sm12-4(122mg)。
esi-ms(m/z):914.5(m+h+)
步骤三:合成产品tm4
在室温条件下,将sm12-4(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品tm4(43mg)。
esi-ms(m/z):814.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.94(s,2h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h)。
实施例5
如图6所示,tm5可通过以下工艺步骤制得:
步骤一:合成中间体sm11-5
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-5(83mg,0.3mmol)溶液1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm11-5(230mg)。
esi-ms(m/z):1150.6(m+h+)
步骤二:合成中间体sm12-5
在室温条件下,将sm11-5(200mg,0.16mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经过制备hplc纯化得到中间体sm12-5(163mg)。
esi-ms(m/z):928.5(m+h+)
步骤三:合成产品tm5
在室温条件下,将sm12-5(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品tm5(31mg)。
esi-ms(m/z):828.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,15h),4.57-4.52(m,2h),4.41(t,1h),4.20-4.10(m,3h),3.30-2.88(m,12h),2.71(d,2h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h)。
实施例6
如图7所示,tm6可通过以下工艺步骤制得:
步骤一:合成中间体sm11-6
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-6(52mg,0.3mmol)溶液1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm11-6(238mg)。
esi-ms(m/z):1150.6(m+h+)
步骤二:合成中间体sm12-6
在室温条件下,将sm11-6(200mg,0.15mmol)加入10ml甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经过制备hplc纯化得到中间体sm12-6(166mg)。
esi-ms(m/z):928.5(m+h+)
步骤三:合成产品tm6
在室温条件下,将sm12-6(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品tm6(56mg)。
esi-ms(m/z):828.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.32-4.10(m,4h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.22(m,8h),0.88-0.81(m,6h)。
实施例7
如图8所示,tm7可通过以下工艺步骤制得:
步骤一:合成中间体sm10-7
在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm10-7(70mg,0.3mmol)溶液1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品过制备hplc得到中间体sm11-7(263mg)。
esi-ms(m/z):1218.7(m+h+)
步骤二:合成中间体sm12-7
在室温条件下,将sm11-7(200mg,0.18mmol)加入10ml30%甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品过制备hplc得到中间体sm12-7(146mg)。
esi-ms(m/z):996.6(m+h+)
步骤三:合成产品tm7
在室温条件下,将sm12-7(100mg,0.12mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品直接过制备hplc,冻干得到产品tm7(53mg)。
esi-ms(m/z):896.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.82(s,2h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),1.22(s,12h),0.88-0.81(m,6h)。
实施例8
如图9所示,tm8可通过以下工艺步骤制得:
步骤一:合成中间体sm10-8
在室温条件下,将乙酸(73mg,0.6mmol),sm9(300mg,0.3mmol)和sm11-8(85mg,0.6mmol)溶液1,2-二氯乙烷(5mlml)中室温反应30min,再加入三乙酰氧基硼氢化钠(256mg,1.2mmol)在室温下反应4h;用30mlml的水淬灭,dcm(30mlml)萃取3次,干燥有机相并旋干得到粗品,粗品过制备hplc得到中间体sm10-8(180mg)。
esi-ms(m/z):1146.6(m+h+)
步骤二、:合成中间体sm12-8
在室温条件下,将sm10-8(160mg,0.14mmol)加入10ml30%甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品过制备hplc得到中间体sm12-8(108mg)。
esi-ms(m/z):924.5(m+h+)
步骤三:合成产品tm8
在室温条件下,将sm12-8(100mg,0.11mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品直接过制备hplc,冻干得到产品tm8(42mg)。
esi-ms(m/z):824.4(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.36-7.12(m,10h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.90-3.46(m,4h),3.30-2.88(m,12h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h),0.71(m,4h)。
实施例9
如图10所示,tm9可通过以下工艺步骤制得:
步骤一:合成中间体sm2-9
在室温条件下,将sm1-9(5g,0.993mmol/g)加入250ml得多肽合成管中,用50ml二氯甲烷溶胀30min,抽干并加入fmoc-d-lys(alloc)-oh(3.34g,7.5mmol)和diea(1.31g,10.0mmol)的dcm(50ml)混合溶液中,室温鼓氮气反应2h后加入5ml甲醇和1mldiea继续反应30min,抽干并用dcm(50ml)洗涤5次。
向得到的产品中加入哌啶/dmf(v/v=1/4,50ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,50ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性。在冰浴条件下分别将fmoc-d-leu-oh(3.35g,10.0mmol),hobt(1.36g,10.0mmol)和hbtu(3.80g,10.0mmol)加入dmf(50ml)活化10min,再加入diea(1.95g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(50ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应。重复以上反应,分别接上两个fmoc-d-phe-oh(3.88g,10mmol)。烘干得到10.82g中间体sm2-9,直接投下一步反应。
步骤二:合成中间体sm3-9
在室温条件下将sm2-9(10.82g)加入三氟乙醇/dcm(100ml,v=/v=1/4)在中,在室温条件下反应2h,抽滤,并用dcm(30ml)洗2次,将有机相浓缩到5ml左右。将浓缩液滴加入100ml的甲基叔丁基醚中搅拌沉降得到5.81g产品sm3-9,lcms(m+h+)=860.4,确认为目标产品。
步骤三:合成中间体sm5-9
在室温条件下,将sm3-9(255mg,0.30mmol),hbtu(172mg,0.45mmol),diea(78mg,0.4ommol0和sm4(50mg,0.33mmol)加入dcm(5ml),在室温条件下搅拌反应4h,lcms检测为目标产品。反应液通过制备hplc纯化得到中间体sm5-9(248mg)。
esi-ms(m/z):999.5(m+h+)
步骤三:合成中间体sm6-9
在室温条件下,将sm5-9(200mg,0.20mmol),pd(pph3)4(23mg,0.02mmol),苯硅烷(20mg,0.40mmol)和dcm(5ml)的混合溶液在室温反应2h,旋干反应液,得到粗品经过制备hplc纯化,冻干得到产品sm6-9(153mg)。
esi-ms(m/z):915.5(m+h+)
步骤四:合成中间体sm8-9
在室温条件下,将乙酸(36mg,0.3mmol),sm6-9(138mg,0.15mmol)和sm7-9(23mg,0.15mmol)溶于1,2-二氯乙烷(5ml)中室温反应30min,再加入三乙酰氧基硼氢化钠(63mg,0.3mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品经过制备hplc纯化得到中间体sm8-9(111mg)。
esi-ms(m/z):1049.6(m+h+)
步骤五:合成产品tm9
在室温条件下,将sm8-9(105mg,0.10mmol)加入加入5ml30%甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到的粗品经过制备hplc纯化得到中间体tm9(36mg)。
esi-ms(m/z):827.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.82-7.12(m,14h),4.57-4.52(m,2h),4.20-4.10(m,3h),3.82(s,2h),3.30-2.88(m,12h),2.72(s,3h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,5h),0.88-0.81(m,6h)。
实施例10和实施例11
如图11所示,tm10和tm11可通过以下工艺步骤制得:步骤一:合成中间体sm10-10在室温条件下,将乙酸(72mg,0.6mmol),sm9(300mg,0.3mmol)和sm11-10(84mg,0.3mmol)溶解于1,2-二氯乙烷(5ml)中,室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml的水淬灭,dcm(30ml)萃取3次,干燥有机相并旋干得到粗品,粗品过制备hplc得到中间体sm10-10(230mg)。
esi-ms(m/z):1267.7(m+h+)
步骤二:合成中间体sm12-10
在室温条件下,将sm10-10(230mg,0.16mmol)加入10ml30%甲胺的甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品过制备hplc得到中间体sm12-7(105mg)。
esi-ms(m/z):1045.6(m+h+)
步骤三:合成产品tm11
在室温条件下,将sm12-10(100mg,0.10mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h,旋干反应液,得到粗品直接过制备hplc,冻干得到产品tm11(45mg)。
esi-ms(m/z):945.6(m+h+)
1hnmr(400mhz,dmso-d6):7.85-7.12(m,10h),4.66-4.54(m,2h),4.22-4.10(m,3h),3.33-2.89(m,12h),2.54(s,3h),2.31-2.17(m,4h),1.66-1.59(m,7h),1.46-1.32(m,15h),0.90-0.81(m,6h)。
步骤四:合成产品tm10
在室温条件下,将tm11(30mg,0.03mmol)加入到naoh(1n,0.3ml)水溶液中,室温反应3h。加入1n盐酸溶液调节ph为6~7,浓缩得到的粗品经过制备hplc纯化得到产品tm10(12mg)。
esi-ms(m/z):834.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.83-7.15(m,10h),4.68-4.54(m,2h),4.23-4.10(m,3h),3.35-2.89(m,12h),2.33-2.16(m,4h),1.65-1.59(m,7h),1.47-1.30(m,15h),0.90-0.80(m,6h)。
实施例12
如图12所示,tm12可通过以下工艺步骤制得:
将无水硫酸镁(10当量)和柠檬酸(5当量)加到tm-10(20mg)的thf溶液中,室温下搅拌过夜。过滤浓缩后粗品经制备hplc纯化后分别得到目标化合物(13mg)。
esi-ms(m/z):990.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.84-7.17(m,10h),4.71-4.56(m,2h),4.25-4.10(m,3h),3.36-2.89(m,12h),2.66-2.16(m,8h),1.65-1.59(m,7h),1.48-1.30(m,15h),0.92-0.81(m,6h)。
实施例13
如图13示,tm13可通过以下工艺步骤制得:
步骤一:合成中间体tm13-1
0℃氮气保护下,将醋酸碘苯(0.3mmol)和2,2,6,6-四甲基哌啶氧化物(0.03mmol)加到sm9(0.3mmol)的二氯甲烷(1ml)溶液中。0℃反应10min后升到室温继续反应30min。tlc监测显示反应完全。反应液用水淬灭并用二氯甲烷萃取。合并所得的有机相用饱和食盐水洗涤、用无水硫酸钠干燥,过滤和浓缩后得到的粗品(320mg)直接用于下一步反应。
esi-ms(m/z):1001.5(m+h+)
步骤二:合成中间体tm13-2
在室温下,将乙酸(72mg,0.6mmol),tm13-1(320mgcrude,0.3mmol)和2-吡咯烷硼酸片哪醇酯盐酸盐(70mg,0.3mmol)溶解于1,2-二氯乙烷(5ml)中,室温反应30min,再加入三乙酰氧基硼氢化钠(126mg,0.6mmol)在室温下反应4h;用30ml水淬灭,然后用二氯甲烷(30ml)萃取3次;合并所得的有机相干燥并浓缩得到的粗品经制备hplc纯化得到中间体tm13-2(158mg)。
esi-ms(m/z):1182.7(m+h+)
步骤三:合成中间体tm13-3
室温下将tm13-2(150mg,0.13mmol)加入10ml30%甲胺甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经制备hplc纯化得到中间体tm13-3(88mg)。
esi-ms(m/z):960.6(m+h+)
步骤四:合成产品tm13
室温下将tm13-2(88mg,0.10mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h。反应液浓缩后的粗品经制备hplc纯化得到产品tm13(31mg)。
esi-ms(m/z):778.6(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.42-7.13(m,10h),4.59-4.52(m,2h),4.20-4.10(m,3h),3.30-2.88(m,8h),2.45-2.30(m,4h),2.29-2.19(m,2h),1.69-1.59(m,6h),1.46-1.32(m,8h),0.88-0.81(m,5h),0.69(brs,2h)。
实施例14
如图14所示,tm14可通过以下工艺步骤制得:
步骤一:合成中间体tm14-1
将碳酸钾(138mg,1.0mmol)加到4-哌啶硼酸片哪醇酯盐酸盐(124mg,0.5mmol)和2-氯乙醛缩二甲醇(62mg,0.5mmol)的四氢呋喃溶液中,室温反应6个h。反应液倒入水中,用二氯甲烷萃取三次,合成所得的有机相用饱和实验室洗涤、经无水硫酸钠干燥后浓缩,得到的粗品(160mg)直接用于下一步反应。
esi-ms(m/z):300.3(m+h+)
步骤二:合成中间体tm14-2
在室温下,将乙酸(60mg,1.0mmol),tm14-1(160mgcrude,0.5mmol)和sm9(500mg,0.5mmol)溶解于1,2-二氯乙烷(5ml)中,室温反应30min,再加入三乙酰氧基硼氢化钠(212mg,1.0mmol)在室温下反应4h;用30ml水淬灭,然后用二氯甲烷(30ml)萃取3次;合并所得的有机相干燥并浓缩得到的粗品经制备hplc纯化得到中间体tm14-2(112mg)。
esi-ms(m/z):1239.7(m+h+)
步骤三:合成中间体tm14-3
室温下将tm14-2(110mg,0.09mmol)加入10ml30%甲胺甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经制备hplc纯化得到中间体tm14-3(57mg)。
esi-ms(m/z):1017.7(m+h+)
步骤四:合成产品tm14
室温下将tm14-3(50mg,0.05mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h。反应液浓缩后的粗品经制备hplc纯化得到产品tm14(13mg)。
esi-ms(m/z):835.6(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.45-7.15(m,10h),4.57-4.52(m,2h),4.21-4.12(m,3h),3.33-2.88(m,8h),2.49-2.30(m,8h),2.26-2.20(m,2h),1.78-1.55(m,8h),1.45-1.32(m,8h),0.89-0.81(m,5h),0.69(brs,2h)。
实施例15
如图15所示,tm15可通过以下工艺步骤制得:
步骤一:合成中间体tm15-1
0℃下将氯乙酰氯(56mg,0.5mmol)滴加到4-哌啶硼酸片哪醇酯盐酸盐(124mg,0.5mmol)和三乙胺(101mg,1.0mmol)的二氯甲烷(5ml)溶液中,室温反应6h。反应液倒入水中,用二氯甲烷萃取三次,合成所得的有机相用饱和实验室洗涤、经无水硫酸钠干燥后浓缩,得到的粗品(160mg)直接用于下一步反应。
esi-ms(m/z):288.2(m+h+)
步骤一:合成中间体tm15-2
将碳酸钾(138mg,1.0mmol)加到tm15-1(160mgcrude,0.5mmol)和sm9(500mg,0.5mmol)的dmf(5ml)溶液中,加热到50℃反应3h。反应液倒入水中,用二氯甲烷萃取三次,合成所得的有机相用饱和实验室洗涤、经无水硫酸钠干燥后浓缩,粗品经制备hplc纯化得到产品tm15-2(133mg)。
esi-ms(m/z):1253.7(m+h+)
步骤二:合成中间体tm15-3
室温下将tm15-2(120mg,0.096mmol)加入10ml30%甲胺甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经制备hplc纯化得到中间体tm15-3(35mg)。
esi-ms(m/z):1030.7(m+h+)
步骤四:合成产品tm15
室温下将tm15-3(30mg,0.03mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h。反应液浓缩后的粗品经制备hplc纯化得到产品tm15(7mg)。
esi-ms(m/z):849.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.43-7.15(m,10h),4.53-4.52(m,2h),4.20-4.10(m,3h),3.36-2.86(m,8h),2.51-2.30(m,6h),2.23-2.20(m,2h),1.81-1.56(m,8h),1.48-1.32(m,8h),0.87-0.81(m,5h),0.71(brs,2h)。
实施例16
如图16所示,tm16可通过以下工艺步骤制得:
步骤一:合成中间体tm16-1
将碳酸钾(138mg,1.0mmol)加到3-氯丙烯基-1-硼酸片哪醇酯(101mg,0.5mmol)和sm9(500mg,0.5mmol)的dmf(5ml)溶液中,加热到50℃反应3h。反应液倒入水中,用二氯甲烷萃取三次,合成所得的有机相用饱和实验室洗涤、经无水硫酸钠干燥后浓缩,粗品经制备hplc纯化得到产品tm16-1(102mg)。
esi-ms(m/z):1168.7(m+h+)
步骤二:合成中间体tm16-2
室温下将tm16-1(100mg,0.09mmol)加入10ml30%甲胺甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经制备hplc纯化得到中间体tm16-2(55mg)。
esi-ms(m/z):946.6(m+h+)
步骤三:合成产品tm16
室温下将tm15-3(40mg,0.05mmol)加入tfa(3ml)和dcm(9ml)的混合溶液中,室温反应0.5h。反应液浓缩后的粗品经制备hplc纯化得到产品tm16(10mg)。
esi-ms(m/z):764.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.46-7.12(m,10h),5.92-5.87(m,1h),5.49-5.43(m,1h),4.59-4.50(m,2h),4.20-4.08(m,2h),3.55-2.85(m,8h),2.45-2.40(m,2h),2.30-2.18(m,2h),1.67-1.55(m,6h),1.45-1.32(m,5h),0.89-0.81(m,6h),0.72(m,2h)。
实施例17
tm17的制备与tm2类似,区别在于,将实施例多肽中间体合成的起始原料fmoc-d-leu-oh替换为fmoc-1-氨基环己烷-1-羧酸。
esi-ms(m/z):826.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.85-7.12(m,14h),4.59-4.52(m,2h),4.23-4.10(m,3h),3.83(s,2h),3.35-2.89(m,12h),2.28-2.19(m,2h),1.66-1.56(m,6h),1.49-1.35(m,6h),0.88-0.80(m,6h)。
实施例18
tm18的制备与tm2类似,区别在于,将实施例多肽中间体合成的起始原料fmoc-d-phe-oh替换为(r)-fmoc-n-(2-苯基丙基)丙氨酸.
esi-ms(m/z):842.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.85-7.11(m,14h),4.58-4.52(m,2h),4.20-4.10(m,2h),3.85(s,2h),3.32-2.88(m,12h),2.27-2.14(m,5h),1.67-1.56(m,6h),1.46-1.30(m,8h),0.88-0.80(m,6h)。
实施例19
如图17所示,tm19可通过以下工艺步骤制得:
步骤一:合成中间体tm19-1
在室温条件下用dcm(20ml)溶胀2-ctcresin(取代度为0.993mmol/g,2.000g),溶胀时间15min,抽掉溶剂,将fmoc-d-lys(boc)-oh(1.120g,2.4mmol)和diea(0.516g,4.0mmol)的dcm(15ml)混合溶液加入溶胀的树脂中,室温条件下反应2h;再加入甲醇(2ml)和diea(1ml),继续反应0.5h;抽干溶剂,再用dcm(30ml)洗三次;最后用dmf(30ml)洗三次后,直接将树脂投下一步反应;
步骤二:合成中间体tm19-2
向步骤一中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-leu-oh(1.809g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性;抽干后直接用于下一步反应;
步骤三:合成中间体tm19-3
向步骤二中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将fmoc-d-phe-oh(1.547g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应;
步骤四:合成中间体tm19-4
向步骤三中得到的产品中加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10min,抽干,再次加入哌啶/dmf(v/v=1/4,20ml),室温条件下反应10mim后抽干,并用dmf(30ml)洗5次,检测最后一次洗涤废液ph为中性;在冰浴条件下分别将boc-d-phe-oh(1.547g,4.0mmol),hobt(0.543g,4.0mmol)和hbtu(1.521g,4.0mmol)加入dmf(20ml)活化10min,再加入diea(0.780g,6mmol)反应5min,最后将活化液加入树脂中,在室温条件下反应2h,用5%的茚三酮显色树脂(在100℃下加热10min),树脂未变色,抽干溶液并用dmf(30ml)洗涤5次,检测最后一次洗涤废液ph为中性,抽干直接用于下一步反应;
步骤五:合成中间体tm19-5
在室温条件下将tm20-4(4.20g)加入三氟乙醇/dcm(50ml,v=/v=1/4)在中,在室温条件下反应2h,抽滤,并用dcm(30ml)洗2次,将有机相浓缩到5ml左右;将浓缩液滴加到100ml的甲基叔丁基醚中搅拌沉降得到1.90g产品.
esi-ms(m/z):860.4(m+h+)
步骤六:合成中间体tm19-6
将tm19-5(0.430g,0.5mmol),hobt(0.135g,1.0mmol),hbtu(0.379g,1.0mmol),和diea(0.202g,2.0mmol)的dcm(15ml)溶液于室温下搅拌0.5h,然后加入2-甲磺酰基-2,7-二氮杂螺[3.5]壬烷(0.150g,0.5mmol)(其合成方法参考专利wo2019015644),室温反应2h。反应液分别用饱和氯化铵溶液、水和饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩后的粗品经制备hplc纯化,得到中间体tm19-6(221mg)。
esi-ms(m/z):1046.5(m+h+)
步骤七:合成中间体tm19-7
向tm19-6(0.200g,0.19mmol)的dcm(10ml)溶液中加入pd(pph3)4(100mg)和苯硅烷(0.218g,2.0mmol),在室温下反应3h,过滤浓缩后得到的粗品经制备hplc纯化,得到中间体tm19-7(105mg)。
esi-ms(m/z):962.5(m+h+)
步骤八:合成中间体tm19-8
在室温下,将乙酸(12mg,0.20mmol),tm19-8(100mg,0.10mmol)和sm10-2(15mg,0.10mmol)溶解于1,2-二氯乙烷(1ml)中,室温反应30min,再加入三乙酰氧基硼氢化钠(42.4mg,0.20mmol)在室温下反应4h;用30ml水淬灭,然后用二氯甲烷(30ml)萃取3次;合并所得的有机相干燥并浓缩得到的粗品经制备hplc纯化得到中间体tm19-8(81mg)。
esi-ms(m/z):1096.5(m+h+)
步骤九:合成中间体tm19
室温下将tm19-8(80mg,0.07mmol)加入10ml30%甲胺甲醇溶液中,在室温条件下搅拌反应1h,直接旋干反应液,得到粗品经制备hplc纯化得到中间体tm19(55mg)。
esi-ms(m/z):874.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.85-7.11(m,14h),4.53-4.50(m,2h),4.20-4.13(m,3h),3.85(s,2h),3.60-2.80(m,16h),2.28-2.19(m,2h),1.75-1.55(m,6h),1.49-1.30(m,5h),0.89-0.80(m,6h)。
实施例20
tm20的制备与tm2类似,区别在于,将实施例多肽中间体合成的起始原料fmoc-d-lys(alloc)-oh替换为tm20-1。
esi-ms(m/z):816.5(m+h+)
1hnmr(400mhz,dmso-d6+d2o):7.80-7.11(m,14h),4.60-4.52(m,2h),4.22-4.10(m,3h),3.85(s,2h),3.32-2.85(m,16h),2.26-2.14(m,2h),1.49-1.31(m,5h),0.89-0.81(m,6h)。
其中,tm20-1的合成路线参考图18:
0℃下氮气保护下,将nah(60%w/w,80mg,2.0mmol)缓慢加入fmoc-d-ser-oh(327mg,1.0mmol)的dmf(5ml)溶液中,0℃反应30min后加入2-溴乙基氨基甲酸烯丙酯(416mg,2.0mmol)。反应体系回到室温继续反应5h后,加入饱和氯化铵溶液淬灭,并用二氯甲烷萃取。合并所得的有机相依次用饱和碳酸氢钠溶液、水和饱和食盐水洗涤,用无水硫酸钠干燥,过滤和浓缩后,粗品经制备hplc纯化得到中间体t20-1(237mg)。
esi-ms(m/z):455.2(m+h+)
其他取代基团的化合物的合成均参考上述实施例中类似合成方法。
对实施例1-9制备的多肽类衍生物进行生物学评价
测试1、对人κ阿片样物质受体的激动效能
1、实验方法:应用稳定表达hkor的细胞株,通过flipr检测方法进行激动剂钙流检测实验。
2、样品检测:每个样品10个检测浓度,复孔;常规检测起点浓度为10um,3倍梯度稀释,384孔板。
3、测试结果:
结论:本发明化合物对人κ阿片样物质受体有明显的激动作用。
测试2、通过测定化合物对dor、kor和mor的抑制作用确定kor选择性
1、实验方法:应用稳定表达人dor、kor和mor的细胞株,通过flipr检测方法进行拮抗剂钙流检测实验。
2、样品检测:每个样品10个检测浓度,复孔。常规检测起点浓度为10um,3倍梯度稀释,384孔板。
3、测试结果:
结论:本发明化合物tm1、tm4、tm5、tm8、tm9和tm15对人κ阿片样物质受体有优异的选择性。
测试3、对细胞色素p450氧化酶的抑制
将含有细胞色素p450的人肝微粒体(0.253mg/ml蛋白)与测试化合物(0.05-50μm)、cyps底物(10μm对乙酰氨基酚、5μm双氯芬酸、30μm美芬妥因、5μm氢溴酸右美沙芬、2μm米达唑仑)、1.0mmnadp在37℃温育10min。将萘黄酮、磺胺苯吡唑、n-3-苄基尼凡、奎尼定、酮康唑作作为参比抑制剂。结果如下表所示,受试化合物的ic50均大于50um。
化合物的cyp450同工酶抑制活性(ic50)
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!