一种碳基增强体/树脂复合材料高强度界面的微波辅助高效构筑方法与流程
本发明属复合材料领域,主要涉及一种碳基增强体/树脂复合材料界面的微波辅助高效构筑方法。
背景技术:
碳基增强体/树脂复合材料由于其高比强度、高比模量、低密度及耐腐蚀性等优异特性,被广泛应用于交通运输、航空航天和体育用品等领域。在碳纤维增强树脂复合材料领域,碳纤维制作的碳化过程导致纤维表面的化学惰性,导致碳基增强体/树脂复合材料中纤维和树脂界面处结合较差,特别在极端条件下在破坏时容易从界面处滑脱,无法充分发挥纤维的强度;碳基纳米粒子增强树脂复合材料中碳基纳米粒子的分散性,与树脂相容性等较难控制,这些限制了碳基增强体/树脂复合材料在一些高性能领域的使用。通常来说,影响碳基增强体/树脂复合材料性能的因素主要有基体,增强体及两者界面。目前碳基增强体/树脂复合材料领域中高强、高模的树脂有相对突出的进展,但是树脂和碳基增强体界面处的结合强度及相容性是目前有待解决的重要问题。目前在碳基增强体改性领域,常用的改性方法为涂敷法、电泳沉积法、化学气相沉积法和化学接枝法。涂敷法相对于其他方法具有方便快捷、易于批量应用的优点,但是过弱的结合力以及涂敷过程中难以控制的均匀性严重降低了其对于界面强度的提升;采用电泳沉积法可以实现上浆层的密实包覆,但是电泳过程中电极气泡易聚集于碳基增强体表面,同时随改性碳基增强体含量增加其电力消耗增加,实际批量改性中性价比较低;化学气相沉积可以通过逐步控制实现纤维等碳基增强体表面改性的均匀性,但是操作复杂,且一般需要贵金属作为表面改性的生长位点,实际改性中性价比较低;化学接枝法可以实现上浆层与碳基增强体界面的较强共价结合,但是增强体表面会受到不同程度的破坏,影响碳基增强体的力学性能。
此外,微波作为具有选择性的电磁复合场在复合材料领域被广泛适用,其特征的选择性加热,快速致热的优势优化了碳基增强体/树脂复合材料的固化过程。同时微波作为辅助场在化学合成领域具有提高反应速率,优化产物质量的优势;微波场具有取向作用,可以诱导电磁响应粒子进行自组装。将纳米粒子改性引入到碳基增强体表面的改性方式本质上包括纳米粒子的化学接枝、物理包覆及其在碳基增强体表面的自组装行为等过程。微波场不仅可以促进纳米粒子接枝的反应效率,还可以诱导纳米粒子在碳基增强体表面的有序自组装。因此微波辅助在碳基增强体/树脂复合材料界面构筑领域具有巨大的潜力。
近年来,微波辅助碳基增强体/树脂复合材料界面增强得到了广泛的研究。在碳基增强体改性方面,yuan等人[compositesscienceandtechnology164(2018)222-228]通过在水氛围中进行微波辐照,可以对碳纤维表面石墨化结构进行破坏,同时在碳纤维表面引入活性基团,提高了碳纤维表面物理黏附和化学活性的双重提升,随后yuan等人[mall2018,14,1703714]发现微波下碳纤维刻蚀碳纤维可实现表面石墨烯量子点的剥离,并且具有明显的微波场下自组装现象。ragoussi等人[j.am.chem.soc.2014,136,4593-4598]对于微波场下氧化石墨烯的电子转移现象,发现在电子转移发生后可以实现酞菁在氧化石墨烯非接枝修饰。在碳基增强体/树脂复合材料界面增强方面,poyraz等人[acsappl.mater.interfaces2015,7,22469-22477]通过界面处二茂铁于微波场下进行损伤焊接,进行界面的快速构筑。menon等人[acsomega2018,3,1137-1146]通过diels-alder点击化学于微波下实现界面的快速修复。但是微波在碳基增强体/树脂复合材料中的应用中存在实际应用的效率较低及可控制性较差等不足之处。综上,微波在碳基增强体/树脂复合材料界面构筑等应用中存在的主要问题如下:一、碳基增强体于微波场中易发生放电现象,实现碳基增强体的改性同时改性程度,均匀度不易控制,易引发爆聚,缺陷和尖端处的放电严重影响碳基增强体/树脂复合材料的界面强度和力学性能;二、微波刻蚀对碳基增强体自身力学性能有一定的损害,碳基增强体的自身结构,缺陷等均影响对于微波的吸收程度,碳基增强体得到活化的同时被刻蚀程度难以控制;三、碳基增强体/树脂复合材料界面传统构筑中,界面上浆剂主要通过粘附力进行界面增强,界面连接作用较弱,并且针对不同的碳基增强体需要的上浆剂差异很大;四、碳基增强体/树脂复合材料界面构筑传统方式过于繁琐,需要长时间多流程的处理,并且对于碳基增强体的性能也有一定的损伤。因此,亟需开发一种对碳基增强体损伤较小、碳基增强体/树脂复合材料界面构筑高效快捷、界面构筑适用性较广的界面构筑方法。
技术实现要素:
一种碳基增强体/树脂复合材料高强度界面的微波辅助高效构筑方法属复合材料领域。碳基增强体/树脂复合材料界面的高效构筑方法包括纳米粒子通过物理包覆和化学接枝协同作用改性碳基增强体,形成笼状结构碳源材料外壳包覆的界面(笼状界面结构)。本发明基于微波辐照活化法,通过调整过渡金属型催化剂、碳源材料与纳米粒子的比例实现碳基增强体活化及纳米粒子的均匀自组装。相较于复合材料传统界面增强更加高效快捷,实现了碳基增强体/树脂复合材料界面的纳米强化,同时解决了在微波场中碳基增强体放电和燃烧破坏其结构完整性和强度的难题,对碳基增强体/树脂复合材料界面的高效构筑具有指导意义。为实现上述目的,本发明提供一种碳基增强体/树脂复合材料高强度界面的微波辅助高效构筑方法,其具体技术内容如下:
本发明中一种碳基增强体/树脂复合材料高强度界面的微波辅助高效构筑方法是通过微波辅助实现碳基增强体的活化以及笼状界面结构在碳基增强体表面的形成,最终形成包覆纳米粒子的的笼状结构的碳源材料外壳包覆的纳米粒子界面结构。
其中,过渡金属型催化剂选自铁、钼、镍、镁中的一种或者几种的盐或金属粉末,可以为其中一种金属组分的单金属催化剂,也可以为两种及以上组成的多金属催化剂,并且所选过渡金属型催化剂均为非负载型金属催化剂;碳源材料选自石墨粉、二茂铁、卟啉中的一种或几种。特殊的,过渡金属型-有机框架化合物可以同时作为过渡金属型催化剂及碳源材料;纳米粒子的结构可以是多维的,包括零维构型纳米粒子(包括二氧化硅微球、四氧化三铁颗粒、碳化硅颗粒、氮化硅颗粒、二氧化钛纳米粒子、酞菁锌微球)、一维构型构型纳米粒子(包括、羧基化碳纳米管、氨基化碳纳米管、酞菁锌纳米线、酞菁镍纳米线、氧化锌纳米棒、碳化硅纳米晶须、硅纳米线)、二维构型纳米粒子(包括石墨烯、二氧化钛纳米薄片、氮化硼纳米片、酞菁纳米薄片)中的一种或几种;碳基增强体选自去浆碳纤维丝束、碳纤维单向复合材料、碳纤维预浸料、碳纳米管薄膜、石墨烯薄膜、富勒烯微球的一种或几种;树脂选自环氧树脂、酚醛树脂、双马来酰胺树脂、聚酰亚胺树脂中的一种或者几种;极性溶剂分为离子型和非离子型,离子型极性溶剂特征为阳离子为季铵盐离子、季鏻盐离子、咪唑盐离子中的一种,阴离子为卤素离子、四氟硼酸根离子、六氟磷酸根离子中的一种;非离子型极性溶剂选自乙二醇、丙酮、n-甲基吡咯烷酮、n,n-二甲基甲酰胺其中的一种或几种的混合,非离子型极性溶剂混合后介电常数需满足公式
本发明目的同时还在于提供了上述微波辅助的碳基增强体/树脂复合材料界面的高效构筑的具体方法:
粉状前驱体及其分散液的制备步骤如下:
1)粉状前驱体的制备:调整过渡金属型催化剂、碳源材料与纳米粒子的质量比例为0.5-1.5:10-100:1-2,充分研磨并且混合均匀制备粉状前驱体,制备的粉状前驱体粒径不超过100nm;
2)粉状前驱体分散液的制备:将充分混合的粉状前驱体1-1.5g与极性溶剂50-100ml使用磁力搅拌在1000-3500rpm的速率下处理1-4h,同时除去未研磨充分的纳米粒子,最终形成粉状前驱体分散液,粉状前驱体分散液于室温下静止10h无团聚现象;
2.包覆纳米粒子的的笼状界面结构的改性碳基增强体制备及其复合材料界面的高效构筑步骤如下:
1)包覆纳米粒子的的笼状界面结构的改性碳基增强体的制备:取粉状前驱体分散液涂覆或浸渍待改性的碳基增强体,控制质量比为0.1-0.5:1-10。随后迅速放置于微波装置中,微波装置频率为2400-2500mhz。根据极性溶剂种类不同加以阶梯微波辐照,第一阶段100-300w处理1-2min引发并进行粉状前驱体与待改性的碳基增强体界面处反应,微波腔内置红外检测装置可观测到包覆纳米粒子的的笼状界面结构的改性碳基增强体的界面处温度升高;第二阶段500-1000w处理0.5-1min完成碳源材料于过渡金属型催化剂下的生长,并且对纳米粒子进行物理包覆和化学接枝,微波腔内置红外检测装置可观测到粉状前驱体与待改性的碳基增强体界面宽度的增加;第三阶段600-800w处理0.5min,通过内置精密天平进行称量,质量浮动于10%内,实现碳基增强体自组装笼状界面结构的形成。
2)包覆纳米粒子的的笼状界面结构的改性碳基增强体/树脂复合材料界面的高效构筑:将包覆纳米粒子的的笼状界面结构的改性碳基增体与所用树脂体系进行复合,其中主体树脂、包覆纳米粒子的的笼状界面结构的改性碳基增强体、固化剂、活性稀释剂质量比例为100:1-10:30-40:20-30;随后采用变功率微波进行阶梯化固化处理,第一段工艺为100-300w下于70-90℃处理15-30min,使树脂体系凝胶;第二段工艺为200-400w下于90-110℃处理20-30min,使树脂体系固化;第三段工艺为400-600w下于120-150℃处理15-25min,使树脂体系固化,最终获得包覆纳米粒子的的笼状界面结构的改性碳基增强体/树脂复合材料。
本发明的效果为:1)纳米粒子作为微波放电电弧的吸收剂和改性剂,解决了在微波场中碳基增强体放电并破坏其结构完整性和强度的难题;2)碳源材料生长的碳纳米管与纳米粒子形成了笼状界面结构,实现了纳米粒子的均匀分散和兼顾物理包覆与化学接枝协同的高强度界面结合,解决了微波辅助改性对于碳基增强体刻蚀所导致的力学性能损失问题;3)提出了微波辅助碳基增强体/树脂复合材料界面构筑的通用方法,解决了传统上浆方法中上浆剂与碳基增强体无法高度匹配的问题;4)该方法操作步骤少,操作安全,具有更高的改性效率,对碳基增强体/树脂复合材料的界面的构筑具有指导意义。
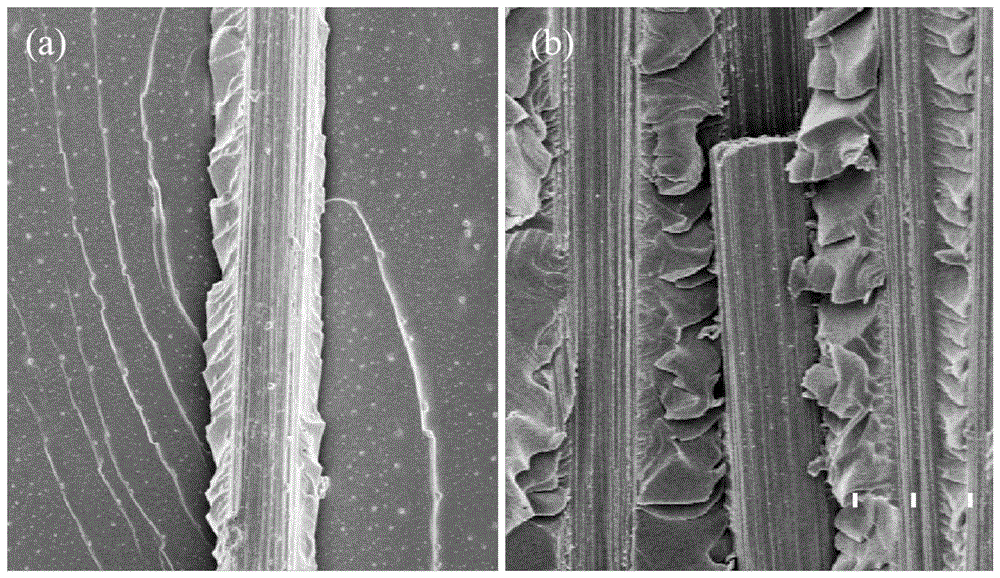
以下结合图表,以具体实施方式对本发明的内容进行详细说明。表1为各实施例中所用到的各具体组分的质量份数,表2为各实施例与对比例中复合材料拉伸强度和复合材料界面剪切强度强度对比,图1为实施例2与对比例2的tfbt测试断面形貌图。
附图说明
图1(a)为实施例2中经过浸渍于氯化亚铁/氧化锌/1-丁基-3-甲基咪唑四氟硼酸盐前驱体溶液中微波辐照处理后碳纤维制备复合材料tfbt断面形貌图;图1(b)为对比例2中未加以微波辐照处理的对比样tfbt断面形貌图。
具体实施方式
本发明采用过渡金属型催化剂、碳源材料和纳米粒子在极性溶剂中的粉状前驱体分散液作为碳基增强体/树脂复合材料界面改性的原料,根据不同类型的碳基增强体以及树脂进行不同过渡金属型催化剂、碳源材料和纳米粒子的搭配,实现碳基增强体/树脂复合材料通过高效快速的微波辅助法引入碳源材料形成的包覆纳米粒子的的笼状界面结构。
其中,其中,过渡金属型催化剂选自铁、钼、镍、镁中的一种或者几种的盐或金属粉末,可以为其中一种金属组分的单金属催化剂,也可以为两种及以上组成的多金属催化剂,并且所选过渡金属型催化剂均为非负载型金属催化剂;碳源材料选自石墨粉、二茂铁、卟啉中的一种或几种。特殊的,过渡金属型-有机框架化合物可以同时作为过渡金属型催化剂及碳源材料;纳米粒子的结构可以是多维的,包括零维构型纳米粒子(包括二氧化硅微球、四氧化三铁颗粒、碳化硅颗粒、氮化硅颗粒、二氧化钛纳米粒子、酞菁锌微球)、一维构型构型纳米粒子(包括、羧基化碳纳米管、氨基化碳纳米管、酞菁锌纳米线、酞菁镍纳米线、氧化锌纳米棒、碳化硅纳米晶须、硅纳米线)、二维构型纳米粒子(包括石墨烯、二氧化钛纳米薄片、氮化硼纳米片、酞菁纳米薄片)中的一种或几种;碳基增强体选自去浆碳纤维丝束、碳纤维单向复合材料、碳纤维预浸料、碳纳米管薄膜、石墨烯薄膜、富勒烯微球的一种或几种;树脂选自环氧树脂、酚醛树脂、双马来酰胺树脂、聚酰亚胺树脂中的一种或者几种;极性溶剂选自去乙二醇、丙酮、n-甲基吡咯烷酮、n,n-二甲基甲酰胺其中的一种或几种的混合,非离子型极性溶剂选自乙二醇、丙酮、n-甲基吡咯烷酮、n,n-二甲基甲酰胺其中的一种或几种的混合,非离子型极性溶剂混合后介电常数需满足公式
下面用实施例对本发明的实施方案进一步说明,但本发明不限于以下实施例。下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
实施例1
将环氧树脂、胺类固化剂、稀释剂的质量份数按照60:37:40的配比制备树脂。环氧树脂采用4,5-环氧己烷-1,2-二甲酸二缩水甘油酯,胺类固化剂采用1,3-二-(γ-氨基丙基)-5,5-二甲基海因,稀释剂采用乙二醇二缩水甘油醚。碳基增强体改性为石墨烯微片,酞菁镍纳米线既作为过渡金属型催化剂又作为纳米粒子。将石墨粉与酞菁镍纳线片分别研磨充分,随后按质量比例为50:1.5进行混合,并且通过双螺旋锥形搅拌机进行充分混合0.5h,使混合纳米粒子外观上不再呈现原纳米粒子成分的状态,形成粉状前驱体。随后将粉状前驱体1.5g与去离子水50ml使用磁力搅拌在3500rpm的速率下处理1h,除去未研磨充分的纳米粒子,形成粉状前驱体分散液。控制粉状前驱体分散液与石墨烯微片的质量比为10:1进行充分浸渍,放入微波装置,所用微波装置具有鼓风设备来稳定温度,并且微波装置频率为2450mhz。随后在微波下进行阶梯处理,第一阶段100w处理3min,第二阶段在600w下处理3min完全挥发溶剂,制备包覆纳米粒子的的笼状界面结构的改性石墨烯微片。将包覆纳米粒子的的笼状界面结构的石墨烯微片与所用树脂体系进行复合,改性的石墨烯微片与树脂体系进行真空脱泡,在密闭舱体和开放环境中对相应树脂体系的微波固化工艺下进行微波固化,第一段工艺为100w下于70℃处理30min,第二段工艺为300w下于100℃处理30min,第三段工艺为600w下于150℃处理30,使树脂体系完全固化固化。试样固化完全后按照相应国标要求打磨平整进行测试。
实施例中具体配方设计如表1中所示。
对比例1
不在环氧树脂上进行粉状前驱体的微波处理,其他条件与实施例1相同由表2可以看出环氧树脂样条在界面处理后拥有更强的拉伸强度,即通过在树脂表面改性使其拉伸性能得到了提升。
实施例2
将东丽t800碳纤维使用丙酮回流去浆24h制备去浆纤维丝束。将氯化亚铁、石墨粉墨与氧化锌纳米线分别研磨充分,随后按质量比例为1:50:0.5进行混合,并且通过双螺旋锥形搅拌机进行充分混合0.5h,使混合纳米粒子外观上不再呈现原纳米粒子成分的状态,形成粉状前驱体。随后将充分混合的纳米粒子1.5g与去1-丁基-3-甲基咪唑四氟硼酸盐50ml使用磁力搅拌在3500rpm的速率下处理1h,同时除去未研磨充分的纳米粒子,制备粉状前驱体的分散液。树脂体系中,主体树脂采用多官能度脂环族环氧树脂与三缩水甘油基三聚异氰酸酯按照质量分数为40:60的复配物,固化剂采用1-氰乙基-2-乙基-4-甲基咪唑与2,2-双(氨基-4-羟基苯基)六氟丙烷按照质量分数为5:10的复配物,活性稀释剂采用乙二醇二缩水甘油醚与己二酸二缩水甘油酯按照质量分数为50:50,主体树脂、固化剂、活性稀释剂三者的质量份数配比为100:30:20。将制备的去浆纤维丝束浸渍于粉状前驱体的分散液充分浸透,随后将浸透完毕的去浆纤维丝束两端使用铝箔封端,迅速放置于微波装置中加以阶梯微波辐照,第一阶段100w处理2min,第二阶段1000w处理1min完成界面处反应并且将溶剂挥发。随后保持张力固定去浆碳纤维,用制备的树脂体系充分浸透去浆碳纤维,固定于tfbt专用聚四氟乙烯磨具中进行微波固化。确定的微波固化工艺定为200w下于80℃处理30min,200w下于100℃处理40min,400w下于120℃处理40min。试样固化完全后按照相应国标要求打磨平整进行测试。
实施例中具体配方设计如表1中所示。
对比例2
不在去浆碳纤维基底上进行纳米粒子的微波处理,其他条件与实施例2相同由表2可以看出改性碳纤维丝束在tfbt界面强度测试拥有更强的拉伸强度,同时fbpo纤维束拔出表现出更强的界面结合强度,图1(a)中实施例2中tfbt测试后力学破坏发生于树脂处,图(b)对比例2中纤维处发生滑移,力学破坏发生于纤维与树脂界面处,表明实施例2中具有更强的界面结合强度,即通过在去浆碳纤维表面改性使其拉伸性能及界面结合性能得到了提升。
实施例3
主体树脂采用三缩水甘油基三聚异氰酸酯与1,3-二缩水甘油基-5,5-二甲基海因型环氧树脂按照质量分数为50:50的复配物,固化剂采用2,2-双(氨基-4-羟基苯基)六氟丙烷与1-氰乙基-2-乙基-4-甲基咪唑按照质量分数为30:10,活性稀释剂采用己二酸二缩水甘油酯,主体树脂、固化剂、和活性稀释剂三者的质量份数配比为100:40:20。酞菁镍纳米线既作为过渡金属型催化剂又作为纳米粒子。将石墨粉与酞菁镍纳米线分别研磨充分,随后按质量比例为50:1.5进行混合,并且充分混合0.5h,使混合纳米粒子外观上不再呈现原纳米粒子成分的状态,制备笼状界面结构前驱体。随后将充分混合的纳米粒子1.5g与n,n二甲基甲酰胺50ml使用磁力搅拌在3500rpm的速率下处理1h,除去未研磨充分的纳米粒子,制备粉状前驱体分散液。控制粉状前驱体分散液与多壁碳纳米管的质量比为15:1进行充分浸渍,放入微波装置,所用微波装置具有鼓风设备来稳定温度,并且微波装置频率为2450mhz。随后在微波下进行阶梯处理,第一阶段200w处理3min,第二阶段在700w下处理3min完全挥发溶剂,制备包覆纳米粒子的的笼状界面结构的多壁碳纳米管。将包覆纳米粒子的的笼状界面结构的多壁碳纳米管与所用树脂体系进行复合,包覆纳米粒子的的笼状界面结构的多壁碳纳米管与树脂体系进行真空脱泡,在密闭舱体和开放环境中对相应树脂体系的微波固化工艺下进行微波固化,第一段工艺为100w下于70℃处理30min,第二段工艺为300w下于100℃处理30min,第三段工艺为600w下于150℃处理30min,使树脂体系完全固化固化。试样固化完全后按照相应国标要求打磨平整进行测试。
实施例中具体配方设计如表1中所示。
对比例3
在环氧树脂底上进行粉状前驱体的分散液的涂敷,但是不进行微波处理,其他条件与实施例3相同由表2可以看出环氧树脂样条在界面处理后拥有更强的拉伸强度,即通过在树脂底表面改性使其拉伸性能得到了提升。
实施例4
将东丽t800碳纤维使用丙酮回流去浆24h制备去浆纤维丝束。将氯化镍、石墨粉墨与四氧化三铁颗粒分别研磨充分,随后按质量比例为0.5:50:0.5进行混合,并且充分混合0.5h,使混合纳米粒子外观上不再呈现原纳米粒子成分的状态,制备粉状前驱体。随后将充分混合的纳米粒子1.5g与n-甲基吡咯烷酮50ml使用磁力搅拌在3500rpm的速率下处理1h,同时除去未研磨充分的纳米粒子,制备粉状前驱体的分散液。树脂体系中,主体树脂采用三缩水甘油基三聚异氰酸酯与1,3-二缩水甘油基-5,5-二甲基海因型环氧树脂按照质量分数为50:50的复配物,固化剂采用2,2-双(氨基-4-羟基苯基)六氟丙烷与1-氰乙基-2-乙基-4-甲基咪唑按照质量分数为30:10,活性稀释剂采用己二酸二缩水甘油酯,主体树脂、固化剂、和活性稀释剂三者的质量份数配比为100:40:20。将制备的去浆纤维丝束浸渍于粉状前驱体的分散液充分浸透,随后将浸透完毕的去浆纤维丝束两端使用铝箔封端,迅速加以阶梯微波辐照,第一阶段100w处理2min,第二阶段1000w处理1min完成界面处反应并且将溶剂挥发,形成包覆纳米粒子的的笼状界面结构的碳纤维。随后保持张力固定去浆碳纤维,用制备的树脂体系充分浸透包覆纳米粒子的的笼状界面结构的碳纤维,固定于专用聚四氟乙烯磨具中进行微波固化。确定的微波固化工艺定为200w下于80℃处理30min,200w下于100℃处理40min,400w下于120℃处理40min。试样固化完全后按照相应国标要求打磨平整进行测试。
实施例中具体配方设计如表1中所示。
对比例4
在去浆碳纤维基底上进行粉状前驱体的浸渍,但是不加以微波处理,其他条件与实施例2相同由表2可以看出改性碳纤维丝束在tfbt界面强度处理后拥有更强的拉伸强度,fbpo纤维束拔出测试中表现出更强的界面结合能力,即通过在去浆碳纤维表面改性使其拉伸性能得到了提升。
实施例5
将通过真空抽滤的氧化石墨烯薄膜作为待改性的碳基底。将氯化钼、石墨粉与四氧化三铁颗粒分别研磨充分,随后按质量比例为1:75:1进行混合,并且通过双螺旋锥形搅拌机进行充分混合0.5h,使混合纳米粒子外观上不再呈现原纳米粒子成分的状态,制备笼状界面结构前驱体。随后将充分混合的纳米粒子1.5g与去丙酮50ml使用磁力搅拌在3500rpm的速率下处理1h,同时除去未研磨充分的纳米粒子,制备粉状前驱体的分散液。树脂体系中,主体树脂采用多官能度脂环族环氧树脂与三缩水甘油基三聚异氰酸酯按照质量分数为40:60的复配物,固化剂采用1-氰乙基-2-乙基-4-甲基咪唑与2,2-双(氨基-4-羟基苯基)六氟丙烷按照质量分数为5:10的复配物,活性稀释剂采用乙二醇二缩水甘油醚与己二酸二缩水甘油酯按照质量分数为50:50,主体树脂、固化剂、活性稀释剂三者的质量份数配比为100:30:20。将制备的氧化石墨烯薄膜浸渍于粉状前驱体的分散液充分浸透,迅速放置于微波装置中加以阶梯微波辐照,第一阶段100w处理2min,第二阶段1000w处理1min完成界面处反应并且将溶剂挥发,制得包覆纳米粒子的的笼状界面结构的氧化石墨烯薄膜。随后用制备的树脂体系充分浸透,固定于专用聚四氟乙烯磨具中进行微波固化。确定的微波固化工艺定为200w下于80℃处理30min,200w下于100℃处理40min,400w下于120℃处理40min。试样固化完全后按照相应国标要求打磨平整进行测试。
实施例中具体配方设计如表1中所示。
对比例5
在氧化石墨烯薄膜基底上进行粉状前驱体的涂敷,不加以微波处理,其他条件与实施例2相同由表2可以看出改性氧化石墨薄膜在tfbt界面强度处理后拥有更强的界面结合强度,即包覆纳米粒子的的笼状界面结构的氧化石墨烯薄膜界面改性使其拉伸性能得到了提升。
表1实施例中各组分的质量份数
表2各实施例与对比例中复合材料拉伸强度和复合材料界面剪切强度对比
- 还没有人留言评论。精彩留言会获得点赞!