一种生产聚乳酸的方法与流程
本发明涉及生物技术领域,具体涉及一种生产聚乳酸的方法。
背景技术:
全球禁塑背景下,生物可降解性生物基材料越来越受到各国政府的重视。到2020年底,我国将率先在部分地区、部分领域禁止、限制部分塑料制品的生产、销售和使用。在此背景下,可降解性聚乳酸(pla)的全球消费量逐年增加,乳酸、聚乳酸行业开始井喷式发展。
聚乳酸是以高光学纯乳酸单体为主要原料聚合得到的一种聚合物。乳酸根据其化学结构的旋光性,分为左旋l型和右旋的d型,自然界中主要存在形式为l-乳酸,d-乳酸由于在人体内无法代谢,其在食品和饮料中的应用受到了限制。乳酸生产方式主要是微生物发酵法,尤其以乳酸细菌为菌种的同型发酵为主,其理论糖酸转化率为100%,即一分子的葡萄糖通过酵解,可以生成两分子的乳酸,发酵过程属于兼性厌氧或者微耗氧状态,过程搅拌和通气水平较为温和。d-乳酸和l-乳酸的生产条件差异主要在菌种及发酵条件,其回收和纯化过程的差异不明显。
在通过微生物发酵法大规模生产乳酸时,为实现产量增加、纯度提高、成本降低、效益提高、菌体耐受性增强等目标,需要进行生物工程上中下游的系统性优化,比如菌株改造、廉价原料替代、生产强度的提升和分离提取的工艺改进,从乳酸生产工艺的多个环节不断挖掘潜力,提高效率。
聚合级高光学纯乳酸原料的聚合工艺相对成熟,改进方向多在工艺参数、产品控制和设备升级等方面,最终实现聚乳酸聚合工艺的提质增效。聚合方法分为以乳酸单体直接脱水缩聚的一步法,及先将乳酸脱水生成丙交酯,再开环聚合制得聚乳酸的二步法。二步法在开环聚合反应时不会产生副产物水,可以精确控制聚合反应的分子量达到10万以上,而且可以在丙交酯的制备纯化上,除去乳酸原料内的杂质及少量的消旋乳酸,提高化学纯度及光学纯度。
但是目前的聚乳酸生产工艺中的各个环节仍有可改善的空间。
技术实现要素:
本发明的目的是为了克服现有技术存在的问题,提供一种生产聚乳酸的方法。
为了实现上述目的,本发明提供一种生产聚乳酸的方法,该方法包括:
(i)将乳酸发酵菌种接种至乳酸发酵培养基中进行发酵,获得含有乳酸根的发酵液;
(ii)对发酵液进行分离处理获得乳酸;
(iii)以乳酸为原料合成聚合级丙交酯;
(iv)聚合级丙交酯在聚合反应装置中经聚合反应得到聚乳酸,其中,所述聚合反应装置包括聚合反应器和设置在所述聚合反应器的流动通道中的搅动组件,所述搅动组件包括电磁绕组机构和磁感应件,所述电磁绕组机构沿所述聚合反应器的内壁环绕设置,所述电磁绕组机构围绕所述磁感应件设置且和所述磁感应件之间形成有间隙,以使所述磁感应件和所述电磁绕组机构能够发生电磁感应,从而使得磁感应件发生绕自身轴线的自转,所述磁感应件上形成有螺纹槽。
通过上述技术方案,本发明能够高效地生产聚乳酸,且在优选的实施方式中,各个环节均能够获得较佳的处理效果。
生物保藏
本发明的鼠李糖乳杆菌cgmccno.19507,于2020年3月25日被保藏在中国微生物菌种保藏管理委员会普通微生物中心(地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮政编码:100101)(保藏单位的缩写为cgmcc),保藏编号为cgmccno.19507。
本发明的鼠李糖乳杆菌cgmccno.19508,于2020年3月25日被保藏在中国微生物菌种保藏管理委员会普通微生物中心(地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮政编码:100101)(保藏单位的缩写为cgmcc),保藏编号为cgmccno.19508。
附图说明
图1是本发明一种实施方式所述搅拌装置的结构示意图;
图2为本发明隔绝空气式丙交酯合成聚乳酸的连续进料系统一实施例中的整体结构示意图;
图3是根据本发明优选实施方式提供的聚乳酸聚合反应装置的示意图;
图4是图3的聚乳酸聚合反应装置的局部示意图,示出了搅拌组件的结构;
图5是本发明制备聚乳酸的装置的示意图;
图6是本发明脱单体反应器iv的示意图;
图7是本发明脱单体反应器iv中的传动装置示意图;
图8是本发明的在线制备改性聚乳酸材料的装置的示意图;
图9是本发明一种实施方式所述的聚乳酸脱挥蒸发器的结构示意图;
图10是本发明另一种实施方式所述的聚乳酸脱挥蒸发器的结构示意图。
附图标记说明
图1中:
1a-圆环形支架,2a-桨叶,3a-轴部,4a-刷式条带
图2中:
1b、原料袋/箱;2b、连接软管;3b、旋风分离器;4b、过滤器;
5b、风机;6b、原料收集器;7b、气体分布器;8b、螺旋输送器;
9b、震动挤压碎料器;a、惰性气体输入管路;b、空分站;c、反应系统
图3-4中:
110-聚合反应器111-熔体进料口112-熔体出料口
120-搅动组件121-磁感应件122-线圈架23-电磁线圈124-隔磁套
131-循环管件132-静态混合器133-循环泵
140-换热液流通腔体141-换热液入口142-换热液出口
图5中:
i、丙交酯熔融罐ii、第一聚合反应器iii、第二聚合反应器
iv、脱单体反应器v、双螺杆挤出机
图6-7中:
4-1、电磁激振器4-2、搅拌驱动电机4-3、连接轴
4-4、聚乳酸熔体入口4-5、导热油入口4-6、导热油出口
4-7、搅拌驱动装置4-7-1、搅拌轴4-7-2、刮板
4-8、丙交酯单体出口4-9、聚乳酸成品出口
图8中:
1c、电机2c、双螺杆挤出机一区
3c、双螺杆挤出机二至三区4c、双螺杆挤出机四区
5c、双螺杆挤出机五区6c、双螺杆挤出机六至十一区
7c、双螺杆挤出机机头8c、第一固体改性剂料斗
9c、第二固体改性剂料斗10c、聚乳酸熔体进料计量泵
11c、第一辅助料斗12c、液体进料计量泵
13c、第二辅助料斗14、聚乳酸熔体管道
图9-10中:
1进料管2分布器
3筒体4夹套
5搅拌轴6搅拌带
7蒸汽凝液出口管8排放管
9锥形筒10出料口
11惰性气体注入管12支撑架
13密封盖14蒸汽入口管
15布料口16密封气体注入管
17密封填料18密封函
19连接杆20支撑杆
21上连接座22加热螺旋盘管
23液态加热介质出口管24液态加热介质入口管
25下连接座26连接座排气孔
27搅拌轴排气孔。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明基于一种或多种乳酸发酵菌种,兼性厌氧条件下同型发酵,得到的发酵液固液分离后,经脱色脱盐处理,并蒸发浓缩得到高纯度高光学纯乳酸原料;基于乳酸原料,乳酸经缩聚反应、丙交酯的纯化等工序得到聚合级丙交酯,聚合级丙交酯经聚合工序、脱单体工序得到聚乳酸熔体,聚乳酸经熔体直纺或切片纺得到聚乳酸纤维,并用于常型和异形纤维的生产。因此,本发明提供一种生产聚乳酸的方法,其特征在于,该方法包括:
(i)将乳酸发酵菌种接种至乳酸发酵培养基中进行发酵,获得含有乳酸根的发酵液;
(ii)对发酵液进行分离处理获得乳酸;
(iii)以乳酸为原料合成聚合级丙交酯;
(iv)聚合级丙交酯在聚合反应装置中经聚合反应得到聚乳酸。
乳酸发酵菌种
可以采用常见的乳酸发酵菌种,包括植物乳杆菌(lactobacillusplantarum)、鼠李糖乳杆菌(lactobacillusrhamnosus)、乳酸片球菌(pediococcusacidilactici),不限于野生菌株、驯化菌株或者经基因工程改造的菌株,上述菌株的一种或多种组合培养;优选为戊糖驯化的乳酸片球菌菌株(如保藏编号为cgmccno.16833的乳酸片球菌)、植物乳杆菌lp-da(cgmccno.16835)、鼠李糖乳杆菌lr-altht(cgmccno.16834)、保藏编号为cgmccno.19507的鼠李糖乳杆菌或保藏编号为cgmccno.19508的鼠李糖乳杆菌。
使用保藏编号为cgmccno.19507的鼠李糖乳杆菌或保藏编号为cgmccno.19508的鼠李糖乳杆菌作为乳酸发酵菌种能够进一步改善发酵效果,提高l-乳酸的产率和光学纯度。
接种至发酵培养基之前,可以先对所述乳酸发酵菌种进行活化,活化的具体方式可以为:以低温冻存的乳酸发酵菌种接种于mrs液体培养基中进行活化培养,温度33-48℃(优选37-42℃),转速150-200r/min,培养过夜制成新鲜种子液。
乳酸发酵培养基
本发明使用的乳酸发酵培养基可以含有碳源、氮源和无机盐。其中,所述碳源可以为被乳酸发酵菌种消耗利用的碳源,包括且不限于葡萄糖、淀粉糖、糖蜜、甘油、蔗糖和纤维素酶解液等原料或其组合。所述氮源可以为被乳酸发酵菌种消耗利用的氮源,包括且不限于酵母粉、酵母膏、玉米浆(粉)、豆粕水解液和棉籽蛋白等原料或其组合。优选地,经过调浆、液化和糖化等预处理过程处理的淀粉糖,或者经粉碎、硫酸酸解、蒸汽汽爆和纤维素酶酶解得到的液相。所述无机盐可以包括缓冲盐、微量元素等,优选地,磷酸缓冲盐、能够提供mg2+和mn2+等的硫酸盐或者盐酸盐。
为了促进乳酸的产生,所述乳酸发酵培养基还可以含有中和剂,所述中和剂为能够中和乳酸、维持发酵液ph的碱性物质,包括且不限于碳酸钙、氢氧化钠、氨水、柠檬酸钠等水解呈碱性的物质或其组合,优选地,氨水或者柠檬酸钠,其中柠檬酸钠类强碱弱酸盐在起到中和作用的同时,可提供额外碳元素,用于生长或产酸。
发酵方法
本发明中,发酵的条件可以包括:按照1-10体积%的接种量,接入新鲜培养的乳酸发酵菌种的种子液,发酵过程温度通常控制在33-48℃,优选37-42℃,发酵转速控制100-300r/min,优选150-250r/min,可开启微量通气为0.05-0.1vvm,控制ph5.5-7,优选ph5.8-6.4。发酵使用的搅拌桨为能够提供理想传质混合效果的搅拌桨,如六平叶搅拌桨、三斜叶搅拌桨、刷式平叶搅拌桨等,优选地,三斜叶搅拌桨与刷式平叶搅拌桨的上下组合。
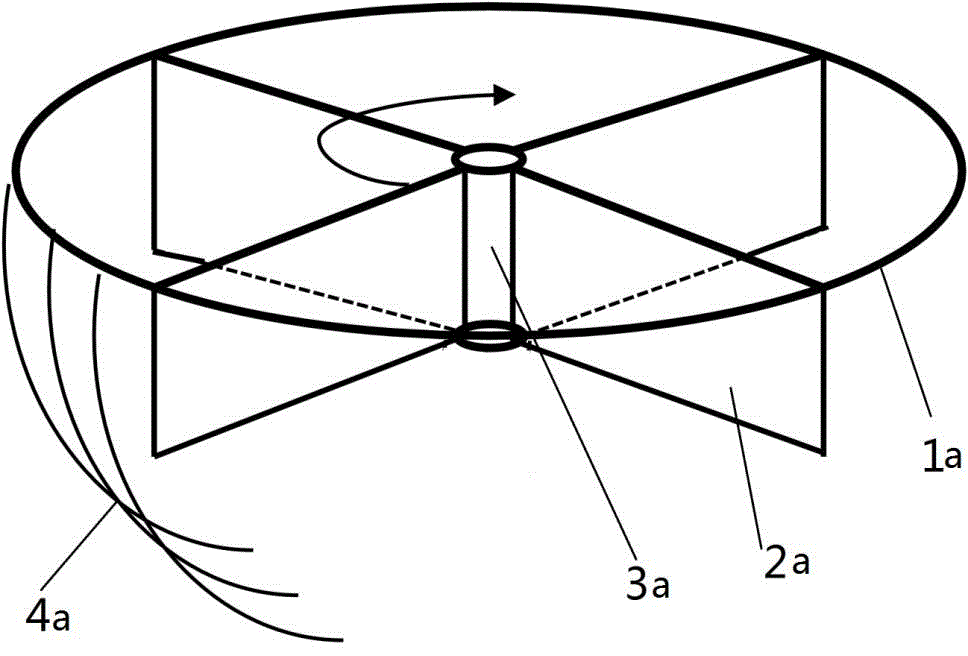
为了克服现有搅拌桨无法搅拌沉淀的问题,如图1所示,本发明提供了一种用于乳酸发酵罐的搅拌装置,包括搅拌桨本体和设置从所述搅拌桨本体垂下的刷式条带4a,以使所述刷式条带4a随着所述搅拌桨本体转动而与所述乳酸发酵罐底部相接触,从而通过所述刷式条带4a扫动和搅拌所述乳酸发酵罐底部的沉淀。
本发明所述的搅拌装置利用刷式条带能扫刷乳酸发酵罐底部的沉淀物,可提升不溶性固体基质在低转速搅拌状态下的混合效果。在乳酸发酵过程中,碳酸钙和乳酸钙沉淀会沉积在发酵罐底部,造成整个反应体系的混合效果下降,对中和能力产生负面影响,借助于该带有刷式条带的搅拌装置,在保证打碎气泡的前提下,将沉积在底部的不溶性钙盐打散,控制乳酸发酵罐内部较为稳定的ph水平,增强整体的混合传质效果。
优选的,所述搅拌桨本体包括轴部3a和设置在所述轴部3a的周向外侧的桨叶2a。现有的搅拌桨多为斜叶搅拌与平叶搅拌的组合或者是平叶搅拌桨,平叶搅拌桨可有效打碎气泡促进氧气的传递,径向混合明显,但轴向混合不充分;而斜叶搅拌与平叶搅拌的组合,可均衡轴向和径向混合,但对于不溶性基质的搅动作用较弱。本发明所述的搅拌桨将原有的圆盘改设为较薄的用于连接转动轴的套筒状,在慢速搅拌状态下,所述桨叶2a与乳酸发酵罐内部的混合溶液的接触面积较大,可以有效地打散气泡,避免乳酸发酵罐中心部位搅拌不均的情况,提高乳酸发酵罐的反应效率。
优选的,所述桨叶2a设置为平板状,所述轴部3a的轴线与所述桨叶2a的板面平行。现有的搅拌桨多为斜叶搅拌与平叶搅拌的组合或者是平叶搅拌桨,平叶搅拌桨可有效打碎气泡促进氧气的传递,径向混合明显,但轴向混合不充分;而斜叶搅拌与平叶搅拌的组合,可均衡轴向和径向混合,但对于不溶性基质的搅动作用较弱。本发明所述的桨叶2a为竖直桨叶,与斜叶桨组合后,在转动速度较慢的状态下,可以有效打碎气泡且使轴向和径向范围内的混合液混合。
优选的,所述搅拌桨本体包括圆环形支架1a,所述圆环形支架1a固定在多个所述桨叶2a的上端外侧,所述圆环形支架1a的中轴线与所述轴部3a的轴线重合。圆环形支架1a和所述桨叶2a相连,进一步增强了搅拌装置的稳定性。
优选的,所述刷式条带4a均匀间隔设置在所述圆环形支架1a圆周边缘。竖直设置的桨叶2a叶片之间能安装所述刷式条带4a位置有限,直接固定于叶片下方,不利于气泡打碎,而圆环形支架1a使得刷式条带可均匀间隔设置,使得刷式条带4a清扫乳酸发酵罐底部更加均匀的同时,也更加均匀的搅拌乳酸发酵罐内的混合液。另外刷式条带4a的安装方式包括:刷式条带4a可以系在所述圆环形支架1a上;刷式条带4a的上端可以贴附在所述圆环形支架1a侧面;所述刷式条带4a上端可以贴附在所述圆环形支架1a底部。
优选的,所述桨叶2a有均匀设置在所述轴部3a周向侧面的3-8片。均匀设置的多个所述桨叶2a能较高效的搅拌乳酸发酵罐内的混合液。
优选的,相邻所述桨叶2a之间的所述圆环形支架1a上设置有1-8根所述刷式条带4a。均匀设置在所述圆环形支架1a上的多个所述刷式条带4a提高了乳酸发酵罐内的混合液的搅拌效率。
优选的,所述刷式条带4a为聚合塑料刷式条带。耐高温耐酸碱的聚合塑料刷式条带有足够韧性,在静止或搅拌状态下,可类似于毛刷一样接触乳酸发酵罐底部,以此提高乳酸发酵罐内混合液中固体小颗粒的混合程度。
本发明还提供一种乳酸发酵罐,包括上述任意一种所述的搅拌装置。本发明所述的搅拌装置利用刷式条带能扫刷乳酸发酵罐底部的沉淀物,可提升不溶性固体基质在低转速搅拌状态下的混合效果。在乳酸发酵过程中,碳酸钙和乳酸钙沉淀会沉积在发酵罐底部,造成整个反应体系的混合效果下降,对中和能力产生负面影响,借助于该带有刷式条带的搅拌装置,在保证打碎气泡的前提下,将沉积在底部的不溶性钙盐打散,控制乳酸发酵罐内部较为稳定的ph水平,增强整体的混合传质效果。
优选的,所述乳酸发酵罐包括反应罐,所述搅拌装置设置在所述反应罐的下部,所述反应罐的底部还设置有环式气体分布器,所述搅拌桨本体的外径大于所述环式气体分布器的外径。在乳酸发酵罐中混合液的反应初期,会有空气由所述环式气体分布器通入所述乳酸发酵罐内混合液中,通入的气泡因浮力一般竖直上升,搅拌桨本体的外径大于所述环式气体分布器的外径使得通入的气泡在所述搅拌桨本体的搅拌范围内,以提高气泡的打散效率,从而提高乳酸发酵罐内混合液接触空气的面积,进一步提高乳酸发酵罐的反应效率。
为了克服d-乳酸发酵生产过程中通气量需求大,设备、能耗以及粮耗高的问题,根据本发明一种具体实施方式,本发明提供一种发酵制备d-乳酸的方法,其特征在于,该方法包括:将植物乳杆菌接种到发酵培养基中进行乳酸发酵,其中,所述发酵培养基中至少部分碳源由糖化液(淀粉糖)提供。通过上述技术方案,可以实现糖利用率和转化率高、生产周期短且原料和生产成本低廉的d-乳酸发酵生产。
其中,淀粉糖:是指利用淀粉质原料(粮食、薯类),经过酸法、酸酶法或酶法制取的糖。包括麦芽糖、葡萄糖和果葡糖浆等,统称淀粉糖。
本发明的发明人在研究的过程中发现,保藏编号为cgmccno.16835的植物乳杆菌在发酵的过程中具有耐抗逆性强,可以耐受较大范围ph和/或温度波动,发酵条件较为宽松。且其对于原料淀粉糖的利用和转化率高,生产周期短,利于工业化生产推广,特别适用于本发明提供的方法。
因此,根据本发明的优选实施方式,其中,所述植物乳杆菌为保藏编号为cgmccno.16835的植物乳杆菌菌株,该菌株已公开于cn109504630a。
根据本发明的优选实施方式,其中,所述淀粉糖的de值为95-99%。
根据本发明的优选实施方式,为了达到以最低廉的原料成本发酵生产d-乳酸的目的,其中,所述方法还包括按照如下方法制备淀粉糖的步骤:
(1)将含淀粉质原料的浆液与淀粉酶混合进行喷射液化,然后闪蒸、降温并保温,得到液化液;
(2)将液化液与糖化酶混合进行糖化,得到糖化液。
根据本发明的优选实施方式,其中,所述含淀粉质原料的浆液中的干物含量为30-50重量%,ph值为5.5-6.5。干物含量指:将测试样品在规定的温度和时间内进行干燥,当干燥至恒重时,干燥后的物质质量占干燥前的样品质量的百分数。
优选地,所述淀粉质原料可以包括谷物原料和/或薯类原料。
更优选地,所述淀粉质原料选自稻谷、玉米、小麦、大麦和高粱中的至少一种。
根据本发明的优选实施方式,其中,相对于每千克以干物计的淀粉质原料,所述淀粉酶的用量可以为100-220u。
更优选地,所述淀粉酶的用量为140-180u。
根据本发明的优选实施方式,其中,所述淀粉酶可以为本领域任意常用淀粉酶,只要能够达到将淀粉质原料中的淀粉水解的目的即可。
优选地,所述淀粉酶可以包括α-淀粉酶、β-淀粉酶和异淀粉酶中的至少一种。
更优选地,其中,为了达到更好的淀粉水解效果,所述淀粉酶可以是α-淀粉酶、β-淀粉酶和异淀粉酶的组合物。
进一步优选地,所述α-淀粉酶、β-淀粉酶和异淀粉酶的重量比可以为1:0.05-0.15:0.1-0.3。特别优选1:0.08-0.12:0.18-0.22。
根据本发明的优选实施方式,其中,所述喷射液化的条件可以包括:温度为100-120℃,时间为1-4s。
根据本发明的优选实施方式,其中,闪蒸的条件可以包括:压力差为(-0.05)-(-0.1)mpa。
根据本发明的优选实施方式,其中,所述降温为喷射所得产物在闪蒸后自然降温5-10℃的操作,以使物料达到保温温度。
根据本发明的优选实施方式,其中,所述保温的方式可以为在80-100℃下保温30-90min。其目的在于使糖化酶在合适的温度下进行反应。
根据本发明的优选实施方式,其中,相对于每千克以干物计的淀粉质原料,所述糖化酶的用量为800-4000u。
更优选地,所述糖化酶的用量为1000-2000u。
根据本发明的优选实施方式,其中,所述糖化的条件包括:温度为55-65℃,ph值为3-5,时间为20-26h。
根据本发明的优选实施方式,其中,所述淀粉糖的用量使得发酵培养基中碳元素的含量为60-80g/l。
根据本发明的优选实施方式,其中,所述发酵培养基中的氮源为玉米浆。所述玉米浆的干物含量为5-45重量%。
根据本发明的优选实施方式,所述发酵培养基还可以含有无机盐。所述无机盐的用量可以为0.25-1g/l。
更优选地,所述无机盐可以包括:磷酸盐、可溶性镁盐和可溶性锰盐的复合物。
根据本发明的优选实施方式,其中,所述发酵培养基中氮源(特别是玉米浆)的用量使得发酵培养基中的氮元素含量为2-3.6g/l。
根据本发明的优选实施方式,其中,所述乳酸发酵的条件可以包括:温度为37-45℃,ph值为5.3-6.5,时间为40-60h,搅拌转速为50-150rpm,相对于所述乳酸发酵体系,所述植物乳杆菌的接种量使得所述植物乳杆菌接种后的生物量od600=0.3-1。
更优选地,所述乳酸发酵的条件包括:温度为37-43℃,ph值为5.5-6.3,时间为40-55h,搅拌转速为80-120rpm。相对于所述乳酸发酵体系,所述植物乳杆菌的接种量使得所述植物乳杆菌接种后的生物量od600=0.3-1。
根据本发明的优选实施方式,其中,在发酵过程中,所述ph的调节方式包括:向发酵体系中加入适量中和剂。
本发明的发明人在研究的过程中发现,石灰乳具有缓释中和的特点,使其作为中和剂应用于乳酸发酵时,可以一次性添加,而不需要在发酵过程中多次添加或持续流加,即可维持合适的发酵液酸碱环境。因此,使用石灰乳作为中和剂可以使乳酸发酵工艺进一步简化。
根据本发明的优选实施方式,其中,所述中和剂可以为石灰乳。相对于乳酸发酵液重量,石灰乳以ca(oh)2计的添加量为8-15重量%(特别优选10.5重量%)。
根据本发明的优选实施方式,其中,所述中和剂还可以包括:氢氧化钠、碳酸钙、氨水、柠檬酸钾和柠檬酸钠中的一种或几种。添加方式为:发酵过程中流加上述中和剂的4-14重量%浓度的溶液,以使得发酵体系保持ph在5.3-6.5范围内。
优选地,所述中和剂为柠檬酸钾和/或柠檬酸钠。
本发明的方法特别适合于在中试或大试水平的乳酸发酵生产。因此,所述方法优选在容积50l以上(特别是50-500l)的发酵罐中进行。
基于纤维素的巨大产能,为了实现乳酸生产低成本高转化率的目的,根据本发明另一种具体实施方式,本发明提供了一种发酵制备乳酸的方法,该方法包括:将乳酸片球菌接种到发酵培养基中,接种后往发酵体系中通入含氧气体进行预发酵,直至发酵体系中乳酸片球菌的浓度达到od600=8-13;然后停止通入含氧气体,继续发酵至发酵体系中的五碳糖消耗量达到45-55重量%,其中,接种前所述发酵培养基中含有30-100g/l的葡萄糖和15-45g/l的五碳糖,所述含氧气体的通气量以氧气计为0.005-0.05vvm。通过上述技术方案,可以将价格低廉,产能巨大的纤维素作为原料应用于乳酸发酵生产中,降低了乳酸发酵生产的成本,并在小试和中试水平验证了本发明提供的方法能够实现低成本高转化的乳酸发酵,为工业化规模乳酸生产工艺提供了参考。单位“vvm”是指l/(l·min)。
根据本发明的优选实施方式,其中,所述乳酸片球菌可以选用保藏编号为cgmccno.16833的乳酸片球菌。该乳酸片球菌已在cn109536409a公开。
接种前所述发酵培养基中除含有五碳糖外,还可以含有40-100g/l的葡萄糖。
根据本发明的优选实施方式,其中,所述发酵培养基中的碳源由纤维素酶解液提供。
优选地,以所述发酵体系为基准,所述发酵培养基中纤维素酶解液的加入量为10-30体积%。
更优选地,所述纤维素酶解液为所述发酵培养基提供25-50g/l的碳元素。
根据本发明的优选实施方式,其中,所述五碳糖包括木糖和/或阿拉伯糖。所述五碳糖可以通过本领域任意常规方式获得,例如可以直接购买商业产品获得,或者通过将任意含五碳糖的原料进行处理获得。
根据本发明的优选实施方式,其中,所述五碳糖由纤维素酶解液提供,所述纤维素酶解液通过将纤维素原料依次进行酸解、汽爆、酶解和固液分离获得。
根据本发明的优选实施方式,其中,所述纤维素原料可以为任意本领域常用纤维素原料。
优选的,所述纤维素原料选自秸秆、玉米芯、硬木、软木、果壳、草、纸、树叶、棉籽絮、柳枝和燕麦壳中的至少一种。
更优选地,所述纤维素原料选择秸秆和燕麦壳中的至少一种。
其中,所述秸秆可以包括任意含纤维素的农作物秸秆。优选为玉米秸秆、小麦秆、棉秆、高粱秆和水稻秸秆中的至少一种。
根据本发明的优选实施方式,其中,所述酸解的方式可以为本领域任意的酸解方式,只要能够达到使木质纤维素结构松散、暴露程度增加的目的即可。
优选地,所述酸解的方式为:将经粉碎的纤维素原料与稀硫酸溶液接触一段时间,然后将所得物料进行脱水得到湿物料。
根据本发明的优选实施方式,其中,所述经粉碎的纤维素原料的粒度可以为10-100mm。所述稀硫酸溶液的浓度为1-2重量%。所述经粉碎的纤维素原料与所述稀硫酸溶液的重量比为0.03-0.08:1。所述接触时间为0.5h以上,优选为0.6-0.8h。
根据本发明的优选实施方式,其中,所述脱水的方式可以为本领域内任意常用脱水方式,只要达到去除接触后物料中的水分的目的即可。
优选地,所述脱水的方式可以为挤压脱水。其优点在于设备简单,降低能耗,一次性脱水处理更为彻底。
根据本发明的优选实施方式,其中,所述汽爆的目的在于实现纤维素、半纤维素和木质素的组分分离。只要能够达到上述目的的任意本领域常用汽爆方式均可适用于本发明提供的方法。
优选地,所述汽爆的方式为:将所述湿物料置于密闭设备中进行蒸汽爆破处理,获得汽爆产物。所述汽爆的条件包括:饱和蒸汽爆破处理的温度为160-170℃,时间为40-60min。
根据本发明的优选实施方式,其中,所述酶解的方式为:将所述汽爆产物与酶进行接触。
优选地,所述酶选自纤维素酶、半纤维素酶,淀粉酶、蛋白酶、葡糖淀粉酶和脂肪酶中的至少一种。
更优选地,所述淀粉酶可以包括α-淀粉酶、β-淀粉酶和异淀粉酶中的至少一种。
更优选地,所述蛋白酶可以包括木瓜蛋白酶、胃蛋白酶和胰蛋白酶中的至少一种。
进一步优选地,为了确保酶解效果,所述纤维素酶可以为纤维二糖酶、半纤维素酶和葡聚糖内切酶组成的复合体系。其中各种酶的配比可以根据纤维素原料的特性不同而进行不同的调整。例如,可以选用市售诺维信公司的cellicctec3纤维素复合酶。
进一步优选地,为了确保酶解效果,所述淀粉酶可以为α-淀粉酶和β-淀粉酶组成的复合体系。其中各种酶的配比可以根据纤维素原料的特性不同而进行不同的调整。例如,可以选用夏盛食品级α-淀粉酶与β-淀粉酶混合制成的复合酶体系,混合比例为1:0.15-0.25(重量比)。
进一步优选地,为了确保酶解效果,所述蛋白酶可以为木瓜蛋白酶、胃蛋白酶和胰蛋白酶的混合物。其中各种酶的配比可以根据纤维素原料的特性不同而进行不同的调整。例如,可以选用市售诺维信公司的protamax复合蛋白酶。
根据本发明的优选实施方式,其中,所述固液分离的原因在于,将上清液作为发酵培养基原料可以减轻后处理负担,从而降低后处理成本。任意本领域常规固液分离方式均可适用于本发明提供的方法。
优选地,所述固液分离的方式包括:板框过滤、离心分离或者自然沉降中的至少一种。
根据本发明的优选实施方式,其中,所述发酵培养基中的氮源由玉米浆提供。
优选地,以所述发酵培养基总体积为基准,其中所述玉米浆的加入量为3-8体积%。
更优选地,所述玉米浆为所述发酵培养基提供0.8-2.3g/l的氮元素。
根据本发明的优选实施方式,其中,所述发酵培养基中还可根据需要添加无机盐。所述无机盐的作用在于维持渗透压、作为功能蛋白酶的激活物质等。
优选地,所述无机盐可以包括:磷酸盐、可溶性镁盐和可溶性锰盐。例如kh2po4、nah2po4、mgso4、mgcl2、mnso4和mncl2等。
更优选地,所述无机盐的添加量为0.25-1.0g/l。
根据本发明的优选实施方式,其中,所述乳酸片球菌的接种方式可以包括:将乳酸片球菌制备成为种子液后接种至培养基中。
优选地,所述种子液的制备方法包括:将低温冻存的乳酸片球菌菌株接种于活化培养基中进行活化培养,过夜培养后制成新鲜种子液。其中,所述活化培养基可以是任意能使低温冻存的乳酸片球菌菌株复苏的培养基。例如,可以是mrs液体培养基和m17培养基中的任意一种。所述活化培养的条件可以包括:温度33-48℃,转速150-200rpm。
更优选地,所述种子液中所述乳酸片球菌的浓度为od600=7-12。
进一步优选地,以所述培养基体积为基准,所述种子液的接种量为1-10体积%。
本发明的发明人在研究的过程中发现,在预发酵过程中适量通入氧气,可以使菌种数量扩增速度加快,从而缩短乳酸发酵周期。
根据本发明的优选实施方式,其中,所述含氧气体可以选用任意包含氧气的气体。
优选地,出于降低成本和便于操作的目的,所述含氧气体为空气。
更优选地,所述含氧气体的通入量以氧气计为0.005-0.05vvm。
根据本发明的优选实施方式,其中,所述预发酵的条件包括:温度为35-48℃,ph值为5.3-6.5,时间为10-30h,搅拌转速为50-200rpm。
更优选地,所述预发酵的条件包括:温度为37-45℃,ph值为5.5-6.2,时间为15-25h,搅拌转速为70-150rpm。
本发明的发明人在研究的过程中发现,当培养基中的五碳糖消耗量达到特定范围时,一定周期内的乳酸的产率达到最高,此时继续进行发酵对于乳酸的产率和生产强度并没有提高。因此,为了避免无意义地延长发酵时间,可以在培养基中五碳糖消耗量达到特定范围时停止发酵,从而缩短发酵周期,提高生产效率。
根据本发明的优选实施方式,其中,所述发酵培养基中的五碳糖含量的检测方式为高效液相色谱法。
根据本发明的优选实施方式,当发发酵体系中的五碳糖消耗量为45-55重量%,乳酸发酵产量达到最大,可以停止发酵。
根据本发明的优选实施方式,其中,所述继续发酵的条件与预发酵的条件可以相同或不同。所述继续发酵的条件包括:温度为37-45℃,ph值为5.3-6.5,时间为30-50h,搅拌转速为50-200rpm。
更优选地,所述继续发酵的条件包括:温度为37-45℃,ph值为5.7-6.2,时间为35-35h,搅拌转速为70-150rpm。
根据本发明的优选实施方式,其中,所述预发酵和/或发酵过程中的ph通过加入中和剂的方式进行调节。根据本发明的优选实施方式,所述中和剂包括:石灰乳、氢氧化钠、碳酸钙、氨水、柠檬酸钾和柠檬酸钠中的至少一种。
优选地,所述中和剂为柠檬酸钾和/或柠檬酸钠。本发明的发明人在研究的过程中发现,将柠檬酸钾和/或柠檬酸钠作为中和剂时,具有在调节ph的同时能够兼顾补充碳源的优点,能够进一步提高乳酸产量。
根据本发明的优选实施方式,其中,出于最大化降低成本和提高糖酸转化率的目的,所述方法可以包括:将乳酸片球菌接种到发酵培养基中,接种后往发酵体系中通入含氧气体进行预发酵,直至发酵体系中的乳酸片球菌的浓度达到od600=10-13;然后停止通入含氧气体,继续发酵至发酵体系中的五碳糖消耗量达到48-53重量%。其中,接种前所述发酵培养基中含有60-80g/l的葡萄糖和25-35g/l的五碳糖,所述含氧气体的通气量以氧气计为0.008-0.05vvm。
根据本发明的优选实施方式,其中,所述方法在中试水平的糖酸转化率可以达到80%以上。其中,所述中试水平为采用100l以上发酵罐进行发酵生产乳酸的水平。
优选地,所述发酵罐具有低剪切、高传质和高混合的特点。
乳酸的分离
可以采用现有的方法从发酵液中分离乳酸,具体包括对发酵液进行固液分离、脱色脱盐处理、蒸发浓缩,从而得到高纯度高光学纯乳酸产品。
固液分离的方式包括但不限于离心、板框过滤、膜过滤、等分离方法。优选板框过滤法,其特征在于,采用气顶水洗法处理过滤结束的滤饼,用压缩空气顶吹出滤饼中的发酵液,空气顶吹结束后使用纯化水浸洗滤饼,浸洗出菌体细胞中残留乳酸。
脱色的方式通常包括但不限于采用活性炭、硅藻土中的一种或两种进行脱色。
脱盐的方式通常包括以下步骤:(1)将离心脱色后的料液通过碱性树脂,除去料液中的大部分阴离子,得到料液i;(2)将步骤(1)中得到的料液i通过酸性树脂,除去料液中的大部分阳离子、氨基酸。
蒸发浓缩的方式通常包括两步蒸发:(1)减压蒸馏除去大部分的水得到料液ii;(2)将料液ii进行分子蒸馏,得到的馏出物即为精制乳酸。
希望说明的是,如果发酵液中的乳酸以乳酸钙的形式存在,则先对发酵液进行酸化以使乳酸钙转化为乳酸,再进行固液分离以除去菌体。如果发酵液中的乳酸以乳酸铵和/或乳酸钠的形式存在,则可直接进行固液分离,后面再进行酸化。
为了克服分离乳酸的工艺比较复杂且制得乳酸纯度比较低的问题,本发明提供一种分离乳酸的方法,该方法包括以下步骤:
(1)将含乳酸溶液(如含乳酸的发酵液)进行离子交换,得到乳酸溶液;
(2)将所述乳酸溶液进行减压浓缩,得到乳酸浓液;
(3)将所述乳酸浓液进行分子蒸馏,得到精制乳酸;
其中,所述含乳酸溶液中,乳酸的含量为5-30重量%。通过上述技术方案,本发明将离子交换、减压浓缩和分子蒸馏进行有效结合,从含乳酸溶液中得到精制乳酸;尤其是采用刮膜蒸发器进行分子蒸馏,得到精制乳酸中,乳酸的含量高达99重量%,有效提高了乳酸的纯度。同时,本发明提供的分离乳酸的方法,简化工艺的流程,便于工业化生产。
在本发明中,对含乳酸溶液具有较宽的选择范围,只要所述含乳酸溶液中,乳酸的含量为5-30重量%即可。优选地,所述含乳酸溶液为乳酸发酵液。一般情况下,所述乳酸发酵液通过使用乳酸发酵菌种进行发酵获得。
如前所述,优选地,所述乳酸发酵菌种选自乳酸球菌、乳酸杆菌、芽孢杆菌和根霉中的至少一种,优选为乳酸杆菌。进一步优选地,所述乳酸杆菌选自鼠李糖乳杆菌、植物乳杆菌和乳酸片球菌中的至少一种,优选为鼠李糖乳杆菌。
根据本发明的一种优选实施方式,所述含乳酸溶液为鼠李糖乳杆菌的发酵液,乳酸的含量为5-30重量%,杂质的含量为7.5-14.4重量%,其中,所述鼠李糖乳杆菌为cgmccno.16834(cn109628339a)。
根据本发明,优选地,所述离子交换在装有离子交换树脂的离子交换系统中进行,其中,所述离子交换系统为本领域的常规技术手段,本发明在此不作赘述。
根据本发明,优选地,所述含乳酸溶液与离子交换树脂的体积比为1:1-10,优选为1:2-5。采用优选的体积比更有利于脱除含乳酸溶液中的阴离子、阳离子和氨基酸等杂质的含量,因此,所述乳酸溶液中,杂质的含量为3-7.5重量%。
优选地,所述离子交换树脂选自阴离子交换树脂和/或阳离子交换树脂。
在本发明中,对所述阴离子交换树脂和阳离子交换树脂的种类和来源具有较宽的选择范围,所述阴离子交换树脂和阳离子交换树脂可以通过商购得到,也可以自制得到;其中,所述阴离子交换树脂可以商购获得,例如:lsd296、lsa-700b和d303中的至少一种;所述阳离子交换树脂可以商购获得,例如:lx-160、lx-732树脂、lx-d001和lx-d151中的至少一种。
根据本发明的一种优选实施方式,将鼠李糖乳杆菌的发酵液(乳酸的含量为14.46重量%)与阴离子交换树脂(浓度)按体积比为1:1-10进行离子交换,得到杂质含量为3-7.5重量%的乳酸溶液。
根据本发明的一种优选实施方式,将鼠李糖乳杆菌的发酵液(乳酸的含量为11.75重量%)与阳离子交换树脂(浓度)按体积比为1:1-10进行离子交换,得到杂质含量为3-7.5重量%的乳酸溶液。
在本发明中,对所述减压浓缩的方式具有较宽的选择范围,只要除去所述乳酸溶液中大部分水,得到水含量为10-40重量%的乳酸浓液即可。优选地,所述减压浓缩在旋蒸仪、升膜蒸发器或降膜蒸发器中进行,优选在旋蒸仪中进行。
根据本发明,优选地,步骤(2)中,所述乳酸浓液中,乳酸的含量为60-90重量%,进一步优选为80-85重量%;水的含量为10-40重量%,进一步优选为15-20重量%。
在本发明中,对所述减压浓缩的条件具有较宽的选择范围,优选地,所述减压浓缩的条件包括:温度为30-80℃,优选为40-70℃,真空度为0.1-1mbar,优选为0.6-0.9mbar。采用优选的减压浓缩的条件更有利于提高乳酸浓液中乳酸的含量。
在本发明中,对所述分子蒸馏的方式具有较宽的选择范围,只要将所述乳酸浓液进行蒸馏,得到高浓度的精制乳酸即可。优选地,所述分子蒸馏在薄膜蒸发器中进行,所述薄膜蒸发器为刮膜蒸发器。本发明实施例中,所述分子蒸馏在刮膜蒸发器中进行,但本发明并不局限于此。
在本发明中,对所述刮膜蒸发器的条件具有较宽的选择范围,优选地,所述刮膜蒸发器的条件包括:刮板转速为20-150r/min,优选为50-120r/min,蒸汽夹套内部温度为80-200℃,优选为100-180℃,真空度为0-1mbar,优选为0.01-0.5mbar。采用优选的刮膜蒸发器的条件,更有利于提高精制乳酸中乳酸的含量。
在本发明中,没有特殊的情况说明下,所述刮膜蒸发器包括加热夹套、内置冷端物料补集器和刮膜器;优选地,所述加热夹套的外侧设有加热介质的出口,所述加热夹套的外侧壁设有加热介质的入口;优选地,所述加热夹套的顶部设有所述乳酸浓液的入口;优选地,所述乳酸浓液进入所述加热夹套进行分子蒸馏后,再沿所述加热夹套内侧壁向下流动进入物料收集器,得到精制乳酸。
根据本发明,优选地,步骤(3)中,所述精制乳酸中,乳酸的含量>90重量%,进一步优选为95-99重量%。
根据本发明,优选地,当所述含乳酸溶液为乳酸发酵液,该方法还包括:在步骤(1)之前,对所述乳酸发酵液进行预处理,预处理的目的在于除去乳酸发酵液中的菌体和色素,从而提高精制乳酸中乳酸的含量。
优选地,所述预处理包括:将所述乳酸发酵液依次进行固液分离和脱色,所述固液分离优选为过滤,所述脱色是将所述乳酸发酵液与脱色剂进行接触。
根据本发明的一种优选实施方式,该方法还包括:在乳酸发酵液进行离子交换前,对所述乳酸发酵液依次进行固液分离和脱色,有利于提高乳酸的纯度,且降低杂质。
根据本发明,优选地,所述脱色剂选自氧化脱色剂和/或吸附脱色剂,优选为吸附脱色剂;进一步优选地,所述吸附脱色剂选自活性炭和/或硅藻土,优选为活性炭。
为了在乳酸盐发酵液菌体细胞分离过程中,克服现有分离菌体细胞与乳酸盐的方法中存在的乳酸盐浪费比较严重和乳酸盐收率较低的问题,本发明还提供了一种分离乳酸盐的方法,该方法包括:
(a)将乳酸盐发酵液与助滤剂依次进行混合、固液分离,得到第一料液和滤饼;
(b)将所述滤饼进行气顶水洗处理,得到第二料液;
(c)将所述第一料液和第二料液的混合液进行下游处理,得到乳酸;
其中,所述乳酸盐选自乳酸铵和/或乳酸钠。本发明采用气顶水洗处理,大幅降低乳酸盐发酵液在固液分离过程中乳酸盐的损失量,提高了乳酸盐的收率;本发明以下提供的分离乳酸盐的系统,简单实用,不需要增加设备投资和试剂耗费,同时降低操作难度,降低乳酸盐生产过程中的单位成本;相比现有技术,乳酸盐的收率≥90%。
在本发明中,对所述乳酸盐发酵液具有较宽的选择范围,只要所述乳酸盐发酵液中乳酸盐的含量为50-300g/ml即可。优选地,所述乳酸盐发酵液通过使用乳酸发酵菌种进行发酵获得,其中,所述乳酸发酵菌种选自乳酸球菌、乳酸杆菌、芽孢杆菌和根霉中的至少一种,优选为乳酸杆菌。
优选地,所述乳酸杆菌选自鼠李糖乳杆菌、植物乳杆菌和乳酸片球菌中的至少一种,优选为鼠李糖乳杆菌。
根据本发明的一种优选实施方式,所述乳酸盐发酵液为鼠李糖乳杆菌的发酵液,乳酸盐的含量为50-300g/ml,其中,所述鼠李糖乳杆菌为cgmccno.16834(cn109628339a)。
在本发明中,为了促进所述乳酸盐发酵液中菌体细胞固液分离,将所述乳酸盐发酵液与助滤剂进行混合,优选地,所述助滤剂选自硅藻土、活性炭和珍珠岩中的至少一种,优选为珍珠岩。
根据本发明,优选地,所述助滤剂与所述乳酸盐发酵液的固液比为(0.1-10):100g/l,其中,固液比是指相对于100l的乳酸盐发酵液,助滤剂的用量为0.1-10g。例如,固液比可以为0.1:100g/l、0.3:100g/l、0.5:100g/l、1:100g/l、2:100g/l、3:100g/l、4:100g/l、5:100g/l、6:100g/l、8:100g/l、10:100g/l以及任意两者之间的中间值,进一步优选为(0.5-5):100g/l,更优选为(0.5-3):100g/l。采用优选的固液比,更有利于降低乳酸盐发酵液中菌体细胞固液分离过程中乳酸盐的损失量,以提分离过程中乳酸盐的收率。
在本发明中,对所述混合的方式具有较宽的选择范围,只要将所述乳酸盐发酵液与助滤剂混合均匀即可。优选地,所述混合的方式选自桨式搅拌和/或框式搅拌,进一步优选为框式搅拌。
优选地,所述混合的条件包括:温度为0-40℃,进一步优选为10-30℃,更优选为15-25℃,转速为10-100rpm,进一步优选为30-80rpm,更优选为40-60rpm。采用优选的混合条件可有效降低乳酸盐发酵液中菌体细胞固液分离过程中乳酸盐的损失量。
根据本发明,优选地,所述固液分离为过滤,本发明中,对所述过滤的方式具有较宽的选择范围,只要将所述乳酸盐发酵液与助滤剂的混合物进行固液分离,得到第一料液和滤饼即可。为了避免乳酸盐发酵液与助滤剂的混合物在菌体分离中乳酸盐的损失,优选所述过滤在板框过滤机中进行。
根据本发明的一种优选实施方式,所述过滤的方式为边搅拌边过滤,也就是说,在过滤过程中,搅拌不停歇。采用这种过滤的方式,可有效促进乳酸盐发酵液的菌体中乳酸盐的溶解,从而降低乳酸盐的损耗。
优选地,所述过滤的压力为0.05-0.5mpa,例如,可以为0.05mpa、0.1mpa、0.15mpa、0.2mpa、0.25mpa、0.3mpa、0.35mpa、0.4mpa、0.45mpa、0.5mpa、以及任意两者之间的中间值,进一步优选为0.1-0.4mpa,更优选为0.15-0.25mpa。
根据本发明,优选地,所述气顶水洗处理的过程包括:将所述滤饼依次进行第一顶吹、水洗和第二顶吹,其中,所述第一顶吹为使用压缩空气将所述滤饼进行第一顶吹,得到乳酸盐透过液;所述水洗为将经过第一顶吹的滤饼用水进行浸泡,得到乳酸盐残留液;所述第二顶吹使用压缩空气将经过水洗的滤饼进行第二顶吹,得到水洗液。
在本发明中,没有特殊的情况说明下,所述第二料液包括所述乳酸盐透过液、乳酸盐残留液和水洗液;其中,所述乳酸盐透过液是指使用压缩空气第一顶吹出滤饼中乳酸盐的透过液,所述乳酸盐残留液是指第一吹顶结束后,使用水浸洗滤饼,浸洗出菌体细胞中乳酸盐的残留液,所述水洗液是指水洗结束后,使用压缩空气第二顶吹出滤饼中乳酸盐的水溶液。
在本发明中,将所述乳酸盐发酵液过滤得到的滤饼进行气顶水洗处理,避免了在过滤过程中,菌体因堆积在滤布表面而导致滤速下降以及在菌体细胞之间的液体中及内部残留乳酸盐,这部分乳酸盐随着菌体细胞的舍弃而损失,造成乳酸盐收率较低。
根据本发明,优选地,所述压缩气体的压力为0.05-0.3mpa,优选为0.1-0.3mpa,更优选为0.12-0.2mpa。
优选地,所述水洗的时间为10-120min,进一步优选为30-90min,更优选为40-80min。
在本发明中,没有特殊的情况说明下,所述下游处理是指将所述第一料液和第二料液的混合液进行浓缩,具体指:先用离子交换脱除阳离子和阴离子,再分子蒸馏除去水分,其中,离子交换和分子蒸馏均为本领域的常规技术手段,本发明对此不作限定。本领域技术人员能够理解的是,所述下游处理还包括进行酸处理使乳酸盐转化为乳酸。
在本发明中,采用上述的分离方法,得到的乳酸盐的收率≥90%,优选≥94%。
本发明还提供了一种分离乳酸盐的系统,该系统包括:依次连通的过滤单元、气顶水洗单元和下游处理单元;
所述过滤单元包括带有搅拌装置的混合罐、液体增压泵、板框过滤机和滤液接收罐;
所述气顶水洗单元包括空气压缩机、压缩空气储罐、水储罐和水输送泵。
根据本发明,优选地,所述板框过滤机中滤布为耐酸型滤布,进一步优选地,所述滤布的目数为200-1000目,优选为300-800目,更优选为400-600目。
在本发明中,采用气顶水洗单元可有效避免乳酸盐发酵液在过滤过程中,因滤饼堆积在滤布上而导致滤速下降缓慢,同时,也避免滤饼中菌体细胞之间的液体以及菌体内部残留的乳酸盐,从而降低乳酸盐的收率。
丙交酯和聚乳酸的制备
基于乳酸原料,通过无接触隔氧进料,乳酸经缩聚反应、丙交酯的纯化等工序得到聚合级丙交酯,再聚合得到聚乳酸。
本发明中以乳酸为原料合成(聚合级)丙交酯优选包括:
(1)在第一聚合条件下,将乳酸进行预缩聚反应,得到聚合度小于5(可以为2、3或4)的乳酸预聚体和含有乳酸的气相;
(2)在第二聚合条件下,在降膜式反应器中,将乳酸预聚体进行缩聚反应,得到聚合度在10以下(可以为5、6、7、8、9或10)的乳酸低聚物;
(3)将所述乳酸低聚物在催化剂的作用下进行解聚,得到含有丙交酯的产物。
为了经聚合反应制备聚乳酸,如图2所示,本发明提供一种隔绝空气式丙交酯合成聚乳酸的连续进料系统,包括原料袋/箱(原料供体)1b和用于收集并输出丙交酯的原料收集器6b,所述原料袋/箱1b与惰性气体输入管路a相连,且所述原料袋/箱1b上可移动地插入一出料管,所述出料管的下游连接有旋风分离器3b,所述旋风分离器3b的固态物质出口与所述原料收集器6b相连。为了方便移动,惰性气体输入管路a和出料管都可以采用连接软管2b,在实际应用中,可以根据需要进行选择。也就是说,本发明在容纳有丙交酯原料的原料袋/箱1b的密闭空间内通过惰性气体输入管路a通入干燥的惰性气体,使所述丙交酯原料与所述干燥的惰性气体形成气固混合物,所述丙交酯原料借助所述惰性气体气动输出,通过旋风分离器3b对输出的所述气固混合物进行气固分离,将分离出来的丙交酯原料收集到原料收集器6b中。由上述内容可知,本发明在隔绝空气和水分的条件下连续进料,应用惰性气体作为推进动力和保护剂,反应的转化率和最终产品的纯度高,损失少,节省人力物力。在实际应用中,惰性气体通常采用氮气。
为了减少丙交酯原料的损失,将旋风分离器3b分离出来的气态物质中混合的少量固态物质进一步分离并收集起来,所述旋风分离器3b的气态物质出口连接有过滤器4b,所述过滤器4b的固态物质出口与所述原料收集器6b相连。为了方便对从旋风分离器3b中分离出来的气态物质进行收集,在本实施例中,过滤器4b采用的是布袋过滤器,在实际应用中,可以根据需要对过滤器4b的形态结构进行选择。为了保持丙交酯原料始终处于与空气和水分的隔离状态,所述惰性气体输入管路a直接与所述过滤器4b相连,以方便对过滤器4b内的物料进行吹扫。
为了使隔绝空气式丙交酯合成聚乳酸的连续进料系统中作为输送动力的气体循环使用,并提高循环过程中气体的流动性,所述过滤器4b的气态物质出口设置有风机5b,风机5b的出口包括两路分支,其中一个分支与所述惰性气体输入管路a相连,另一个分支与设置在所述隔绝空气式丙交酯合成聚乳酸的连续进料系统外部的空分站b相连。也就是说,在旋风分离器3b中进行气固分离后输出的气体还要进入过滤器4b进行过滤,将过滤出来的丙交酯原料固体收集到原料收集器6b中;过滤后的气体中的一部分惰性气体返回惰性气体输入管路a,重新输送到原料袋/箱1b中,形成惰性气体在隔绝空气式丙交酯合成聚乳酸的连续进料系统中的循环使用;另一部分惰性气体输出另行回收处理,比如:在本发明的一个实施例中,另一部分惰性气体输出至空分站b进行回收处理,上述两部分惰性气体按照一定的比例进行分配。
为了防止原料收集器6b中收集的丙交酯原料发生聚集、粘连成块,所述原料收集器6b内设有用于使容置在所述原料收集器内的固态物质松动的气体分布器7b,气体分布器7b直接与惰性气体输入管路a相连,通过惰性气体将收集好的丙交酯进行松动。
为了方便转运,所述原料收集器6b的出口与螺旋输送器8b相连,所述螺旋输送器8b的出口与设置在所述隔绝空气式丙交酯合成聚乳酸的连续进料系统外部的反应系统c相连。
通常情况下,所述惰性气体可以为氮气、氩气或氦气。处于成本和操作方便性考虑,在本实施例中,采用的是氮气。
另外,根据需要,所述原料袋/箱1b的结构也可以采用多种形式,比如:可以为封闭的箱体或袋体,在本实施例中,采用的是封闭箱体。由于丙交酯原料可以成箱包装也可以成袋包装,因此氮气管路和丙交酯出料输送管路可以由软管进行连接,鉴于连接软管更加方便移动,这样无论哪种结构形式的包装,都可以通过插入包装箱或包装袋中的方式实现对丙交酯原料的气动输送。进一步地,为了防止丙交酯原料的原料包装中受潮结块从而影响进料,还可以在原料袋/箱1b的外部设有震动挤压碎料器9b,在进料过程开始之前,先对受潮结块的丙交酯原料进行充分地物料挤压震动,将在原料包装中结块的所述丙交酯原料充分打散。
结合图2所示,本发明还提供一种如上所述的隔绝空气式丙交酯合成聚乳酸的连续进料系统的进料方法,包括如下步骤:
步骤100:在容纳有丙交酯原料的密闭空间内通入干燥的惰性气体,使所述丙交酯原料与所述干燥的惰性气体形成气固混合物,所述丙交酯原料借助所述惰性气体气动输出;
步骤200:对输出的所述气固混合物进行气固分离,将分离出来的丙交酯原料收集并输出至设置在外部的反应系统。
所述步骤100中的所述干燥的惰性气体中的水分含量为:15ppm(v/v)以下;氧气含量为:50ppm(v/v)以下。
为了确保丙交酯原料始终处于与空气和水分的隔绝状态,所述步骤100之前包括:在所述隔绝空气式丙交酯合成聚乳酸的连续进料系统首次使用时,先采用干燥的惰性气体对整个进料系统中残留的空气进行吹扫置换,直到进料系统内惰性气体的含量超过99%(v)。
为了防止丙交酯原料的原料包装中受潮结块从而影响进料,所述步骤100之前包括:对所述丙交酯原料进行充分地物料挤压震动,将在原料包装中结块的所述丙交酯原料充分打散。所述物料挤压震动的处理时间为:5-30min;优选为:5-20min。
为了节约能源,使惰性气体在整个进料系统中能够反复使用,形成惰性气体在隔绝空气式丙交酯合成聚乳酸的连续进料系统中的循环,具体来说,所述步骤200进一步包括:对进行气固分离后输出的气体进行过滤,收集过滤出来的所述丙交酯原料固体;过滤后的气体中的一部分惰性气体返回所述步骤100中容纳有所述丙交酯原料的密闭空间内,形成惰性气体在隔绝空气式丙交酯合成聚乳酸的连续进料系统中的循环使用;另一部分惰性气体输出另行回收处理,比如:可以输出至空分站进行回收处理。更具体地,循环使用的所述一部分惰性气体和输出至空分站进行回收处理的所述另一部分惰性气体的分配比例为:5:1-1:2,优选为:4:1-1:1。
对收集好的所述丙交酯原料还需要进行定期惰性气体吹扫;所述定期惰性气体吹扫的频率为每完成一个批次的进料后进行一次;所述批次为:每完成4-6个丙交酯原料密封包装量的进料为一个批次。
另外,为了防止收集好的丙交酯原料发生聚结影响固态物料输送,所述步骤200进一步包括:对收集好的所述丙交酯原料固体进行惰性气体送风松动处理,所述送风松动的时间为:5-30min,优选为:5-20min。
在确保空气和水气完全隔离的情况下,隔绝空气式丙交酯合成聚乳酸的连续进料系统的环境温度为:20-35℃。
由上述内容可知,本发明提供一种隔绝空气式丙交酯合成聚乳酸的连续进料系统,应用惰性气体,比如:氮气,作为丙交酯原料运输的推进动力和转运过程中的保护剂,既可以隔绝空气和水分进入系统,保证丙交酯的纯度,从而提高反应的转化率和最终产品的纯度,又可以避免原料的损失,减少系统内的无用废料的产生,同时减少人工操作,节省人力物力。
结合图2所示,本发明所提供的隔绝空气式丙交酯合成聚乳酸的连续进料系统的具体工作过程是这样的:
成箱密封包装有原料丙交酯的原料袋/箱1b移送至原料处理台并固定,在如图2所示的实施方式中,震动挤压碎料器9b主要包括设置在原料处理台的震动机构,通常可以选择为震动弹簧。通过电机等驱动机构驱动原料处理台上下、左右单向震动或上下左右组合震动,将在原料包装中结块的所述丙交酯原料充分打散。通常情况下,震动挤压碎料器9b对所述丙交酯原料进行充分地物料挤压震动时间为5-30min,优选5-20min,比如:在本实施例中物料挤压震动10min,即可将在原料包装中结块的所述丙交酯原料充分打散。从惰性气体输入管路a中输出的惰性气体,通过一根连接软管2b所连接的可移动的惰性气体输入管路,插入到原料袋/箱1b的底部与其内部连通,在本实施例中采用的惰性气体为氮气,同时将另一根连接软管的可移动的原料出料管的一端也插入到原料袋/箱1b中与其内部连通,原料出料管的另一端与旋风分离器3b内部连通,且在原料出料管上设有用于控制该管路开闭的控制阀。通常情况下,输入的氮气为干燥的氮气,其中的水分含量为15ppm(v/v)以下,氧气含量为50ppm(v/v)以下,在本实施例中水分含量为10ppm(v/v),氧气含量为40ppm(v/v)。
需要说明的是,在第一次开启系统时,需要先用新鲜、干燥的氮气对整个系统的空气进行吹扫置换,直到系统内的氮气含量超过99%(v)时,将氮气管和出料管插入到原料袋/箱1b里,同时开启风机5b对丙交酯原料进行气动输送。
开启氮气管上的氮气进料阀,氮气带动丙交酯经出料管输送至旋风分离器3b,在旋风分离器3b里丙交酯与氮气分离,丙交酯落至旋风分离器3b的底部,氮气自旋风分离器3b的顶部至过滤器4b。
落到旋分分离器3b的丙交酯原料经底部出料口进入原料收集器6b,然后再由螺旋输送器8b送至反应系统c。原料收集器6b底部设有气体分布器7b,在丙交酯原料堵塞时可通入氮气对丙交酯原料进行松动,以保证原料输送顺畅,每次松动用时5-30min,优选5-20min,比如:在本实施例中,送风松动10min。此外,还需要对原料收集器6b中收集好的所述丙交酯原料进行定期惰性气体吹扫,所述定期惰性气体吹扫的频率为每完成一个批次的进料后进行一次;其中,每完成4-6个丙交酯原料密封包装量的进料为一个批次,在实际操作过程中可以根据需要对吹扫的频率进行选择。在本实施例中,对原料收集器6b中收集好的丙交酯原料进行定期氮气吹扫,也是通过气体分布器7b来完成的。
气固分离后输出的气体通过过滤器4b进行过滤,过滤出来的所述丙交酯原料固体在原料收集器6b中收集;过滤后的氮气一部分返回原料袋/箱1b内,形成氮气在隔绝空气式丙交酯合成聚乳酸的连续进料系统中的循环使用;另一部分氮气输出至空分站b进行回收处理。通常情况下,一部分和另一部氮气的分配比例为:5:1-1:2,优选为:4:1-1:1,在本实施例中,进入过滤器4b后的氮气携带的丙交酯原料在过滤器4b中被收集下来,氮气被风机5b抽送出去后,80%(v)的氮气返回到原料袋/箱1b处的氮气管路来代替新鲜氮气对丙交酯原料进行气动输送,其余20%(v)的氮气送至空分站b进行再生处理。也就是说,在本实施例中,一部分和另一部分氮气的分配比例为:4:1。
在上述的进料过程中,隔绝空气式丙交酯合成聚乳酸的连续进料系统的环境温度为:20-35℃,比如:在本实施例中,环境温度为25℃。
由本发明所提供的隔绝空气式丙交酯合成聚乳酸的连续进料系统的上述具体工作过程可知,在图2所示的实施例中,整个系统在每次运行过程中只需要补充少部分新鲜氮气,大部分氮气可以循环利用,节约能耗。同时整个系统密闭无空气接触,可以保证原料丙交酯的纯度,同时避免原料损失。
在此进料系统后,应用如下工艺以丙交酯为原料合成聚乳酸,合成聚乳酸的反应全过程都在高真空或者氮气保护下进行,具体包括:
(a)在氮气保护下将丙交酯通过螺旋进料器送入丙交酯熔融罐中进行熔融处理,得到熔融态丙交酯,融化温度90-110℃,反应时间1-1.2h。
(b)在催化剂和引发剂的存在下,将所述熔融态丙交酯和复合稳定剂在第一聚合反应器中进行第一聚合反应,得到第一熔体,反应温度140-160℃;压力为50-53kpa,反应时间为3-3.2h;
(c)将所述第一熔体通入第二聚合反应器中进行第二聚合反应,得到第二熔体;反应温度为170-200℃,压力为6-6.5mpa,反应时间为1-1.2h;
(d)将所述第二熔体通入脱单体反应器中进行脱单体处理;温度为210-215℃,压力为1-1.5kpa,反应时间为0.5-0.6h;搅拌速率为5-30rpm;
(e)步骤(d)后得到的聚乳酸熔体进行水冷切粒、脱水、结晶和干燥处理,最终得到聚乳酸树脂,造粒机温度为210℃。
通过上述实际的工业化生产并对采用本发明所提供的进料系统和方法生产出来的聚乳酸的反应转化率高,可达97%以上;所得聚乳酸产品色泽度好,重均分子量为13万-25万;所得聚乳酸中丙交酯单体的含量为0.7-2.0‰;所述聚乳酸在2.16kg载荷下190℃的熔融指数6-27g/10min。与现有技术中反应转化率95%左右,单体含量2-5‰的情况相比,本发明应用惰性气体作为推进动力和保护剂,既可以隔绝空气和水分进入系统,避免了丙交酯原料因吸湿和氧化引起的变质,又提高了反应转化率和最终产品纯度,反应转化率可达97%,且最终产品中单体含量可降低至0.7-2.0‰;同时,本发明还能避免原料的损失,减少系统内的无用废料的产生,因操作简单而减少人工,节省人力物力,是一种可工业化的连续进料方式。
通过上述技术方案,本发明在隔绝空气和水分的条件下连续进料,应用氮气作为推进动力和保护剂,避免了丙交酯原料因吸湿和氧化引起的变质,提高了反应转化率和最终产品纯度,本发明中反应转化率可达97%,最终产品中单体含量可降低至0.7-2.0‰;且本发明操作简单,节省人力物力,是一种可工业化的连续进料方式。
本发明涉及一种聚乳酸聚合反应装置,所述聚乳酸聚合反应装置包括聚合反应器110和设置在所述聚合反应器110的流动通道中的搅动组件120,所述搅动组件120包括电磁绕组机构和磁感应件121,所述电磁绕组机构沿所述聚合反应器110的内壁环绕设置,所述电磁绕组机构围绕所述磁感应件121设置且和所述磁感应件121之间形成有间隙,以使所述磁感应件121和所述电磁绕组机构能够发生电磁感应,从而使得磁感应件121发生绕自身轴线的自转,所述磁感应件121上形成有螺纹槽。
在泵件的动力作用下,熔体会进入聚合反应器110并最终从聚合反应器110中输出,在聚合反应器中的温度和压力条件下发生聚合反应,本申请的搅动组件120设置在聚合反应器110的流动通道中,磁感应件121和电磁绕组机构之间会发生电磁感应,使得磁感应件121能够设置在电磁绕组之间,其中在电磁绕组机构的通电状态和断电状态下,磁感应件121均能够发生绕自身轴线的自转,由于熔体需要经由磁感应件121和电磁绕组机构的间隙,而磁感应件121的表面上具有螺纹槽,因此熔体会被磁感应件搅动,配合熔体自身的流动压力,熔体会做周期性的轴向脉动,从而实现熔体周期性体积脉动形变输运,保证分子量均匀性,聚乳酸熔体在脉动形变挤出过程中,熔体停留时间缩短,物料不易分解,从而提高了聚合反应的效果,使得产品的转化率得到提高。
其中,如图3所示,聚合反应器110为圆柱状且沿竖直方向设置,磁感应件121和聚合反应器110的轴线共线,聚合反应器的底部设置有熔体进料口111,底部设置有熔体出料口112,在优选的实施方式中,搅动组件120设置在更靠近底部的熔体进料口111的位置,比如设置搅动组件120的中心处和熔体进料口111之间的距离为聚合反应器110的总长度的三分之一。
在聚合反应器110的内部设置有主轴杆,主轴杆沿竖直方向延伸,磁感应件121套设在主轴杆上。其中在一种优选实施方式中,磁感应件121通过轴承套设在主轴杆上,且轴承的内环和主轴杆形成有比如通过螺栓固定的连接形式,轴承的外环能够支撑磁感应件121从而使得磁感应件121保持在环形的电磁绕组机构中,磁感应件121转动时能够相对于主轴杆发生转动,即磁感应件转动而主轴杆不动;而在另一种优选实施方式中,主轴杆可转动地设置在聚合反应器110中,磁感应件121固定设置在主轴杆上,当磁感应件121转动时,磁感应件121和主轴杆共同转动。
如图4所示,电磁绕组机构包括线圈架122和缠绕设置在线圈架122上的电磁线圈123,其中,线圈架122为和聚合反应器110的内径相匹配的圆环状,可以通过螺栓、螺钉连接或者粘接的方式固定在聚合反应器的内壁上。磁感应件121包括沿溶体流动方向依次设置的主体部和尖端部,所述主体部为圆柱状,所述尖端部为锥状且沿溶体流动方向的直径逐渐减小,且尖端部突出于所述电磁绕组机构设置。其中圆柱状的主体部使得磁感应件121在转动时能够充分地对经过间隙的熔体进行搅动,而突出于线圈架122的尖端部能够保证熔体从间隙排出之后的流动性。其中磁感应件121为一体成型件并选用38crmoal为材料制作,且磁感应件为实心件。
聚合反应器外部安装有电控制器,电控制器通过电线向电磁线圈供电且能够控制电路的连通和断开,电线可以穿过聚合反应器的外壳从而和电磁线圈电连接,也可以在聚合反应器的外壳上设置导电螺栓,利用导电螺栓向聚合反应器中的电磁线圈供电。
此外,为了避免磁感应件和电磁线圈直接接触而造成短路,在电磁绕组机构的外侧还设置有隔磁套124,隔磁套124位于磁感应件121和电磁线圈123之间,隔磁套的材料为高导磁材料,磁导率在80000-350000之间,隔磁套能够直接套设在电磁绕组机构上。
为了保证聚合反应器的换热效果,如图3所示,聚合反应器110包括包括外壳体和内壳体,所述外壳体和内壳体之前形成有换热液流通腔体140,所述外壳体上设置有与所述换热液流通腔体140连通的换热液入口141和换热液出口142,所述搅动组件120设置在所述内壳体中。通过换热液入口141和换热液出口142可以向换热液流通腔体140通入换热液,使得换热液能够和内壳体中的熔体进行热交换,使得熔体的温度升高。
更优选的,如图3所示,本申请提供的聚乳酸聚合反应器还包括循环管件131和设置在所述循环管件131上的静态混合器132、循环泵133,其中循环管件131的两端分别流体连通所述聚合反应器110的位于所述搅动组件120的两侧的部分。在一批熔体从熔体进料口111进入聚合反应器110的内部之后,关闭熔体进料口111并保持熔体出料口112关闭,此时在循环泵133的作用下,熔体会在聚合反应器110和循环管件131之间循环流动,且每个循环都会经过静态混合器132,静态混合器的内部具有多个交替排列的长方形挡板,长方形挡板上有通孔,比如直径10cm圆孔,实现了对熔体适当的阻挡,保证助剂和熔体混合均匀,从而提高熔体与助剂的混合效率,进一步提高聚合效果。
熔体可以在聚合反应器110和循环管件131上进行多个循环流动,通过控制搅动组件的电磁绕组机构是否通电,能够控制磁感应件121的自转方向,在电磁绕组机构通电和非通电的状态下,磁感应件121的自转方向是正好相反的,在使用中,可以将电磁绕组机构保持通电一段时间,之后再保持电磁绕组机构不通电一段时间,使得熔体能够得到不同方向的搅动,提高聚合反应的效果。
将融态的丙交酯,催化剂、引发剂、高效复合稳定剂通入该聚合反应器,在压力6mpa,温度190~200℃的情况下,以熔体在聚合反应器110和循环管件131之间反应时间3小时为例,其中在通电状态下向搅动组件通入的电流为20-30之间,以搅动组件通电和断电的时间为变量得到表i中的结果。
表i
从表i中可以看到,利用了本申请提供的聚乳酸聚合反应装置生产的pla切片多分散指数(pdi)最小为1.15,而现有常规聚合工艺生产的pla切片(pdi)为1.56。
本申请还提供一种聚乳酸聚合反应系统,聚乳酸聚合反应系统会进行两次聚合反应工序,本申请提供的聚乳酸聚合反应装置用于系统的第二次聚合反应工序,第一次聚合反应之后的熔体会从熔体进料口111进入到聚合反应器110中,并经过搅动组件120的搅动处理,之后经由熔体出料口112输出,进入下游以继续执行脱单体工序以及造粒工序。本申请提供的聚乳酸聚合反应系统能够提高聚合反应的效果,使得产品的转化率得到提高。
通过上述技术方案,利用本申请提供的聚乳酸聚合反应装置,在泵件的动力作用下,熔体会进入聚合反应器并最终从聚合反应器中输出,在聚合反应器中的温度和压力条件下发生聚合反应,搅动组件设置在聚合反应器的流动通道中,磁感应件和电磁绕组机构之间会发生电磁感应,使得磁感应件能够设置在电磁绕组之间,通过对电磁绕组机构的进行供电和断电,即电磁绕组机构在通电状态和断电状态下磁感应件均能够发生绕自身轴线的自转,由于熔体需要经由磁感应件和电磁绕组机构的间隙,而磁感应件的表面上具有螺纹槽,因此熔体会被磁感应件搅动,配合熔体自身的流动压力,熔体会做周期性的轴向脉动,从而实现熔体周期性体积脉动形变输运,保证分子量均匀性,聚乳酸熔体在脉动形变挤出过程中,熔体停留时间缩短,物料不易分解,从而提高了聚合反应的效果,使得产品的转化率得到提高。
为了经聚合反应制备聚乳酸,本发明提供了一种生产聚乳酸的装置,其中,所述装置包括依次连接的丙交酯熔融罐i、第一聚合反应器ii、第二聚合反应器iii和脱单体反应器iv;其中,所述脱单体反应器iv包括电磁激振器4-1、搅拌驱动电机4-2和搅拌驱动装置4-7,且所述搅拌驱动电机4-2与所述搅拌驱动装置4-7相连接以对所述搅拌轴4-7-1的搅拌频率进行调控,以及所述电磁激振器4-1通过振动传递盘与所述搅拌驱动电机4-2相连接以使所述搅拌驱动电机4-2进行轴向振动。
在本发明中,图7是本发明脱单体反应器iv中的传动装置示意图,如图7所示,在所述脱单体反应器iv中,所述电磁激振器4-1依次与所述搅拌驱动电机4-2和所述连接轴4-3相连接设置,所述电磁激振器4-1通过振动传递盘与所述搅拌轴4-7-1相连接以使所述搅拌轴4-7-1进行轴向振动,所述搅拌轴4-7-1上的所述刮板4-7-2也随着所述搅拌轴4-7-1振动发生周期性变化,进而能够将振动力场引入所述脱单体反应器iv中的熔体(例如,经第二聚合反应后得到的第二熔体)中,更进一步地,熔体的剪切速率随振动力场也作周期性变化,振动力场的引入能够提高熔体剪切速率,使熔体界面拉伸、压缩,产生相应的振荡。
根据本发明,图6是本发明脱单体反应器iv的示意图,如图6所示,所述搅拌轴4-7-1上设置有多个刮板4-7-2;优选地,在以所述搅拌轴4-7-1上任一点为中心的水平面上,多个所述刮板4-7-2以36°-180°角度间隔地沿所述搅拌轴4-7-1的周向设置,具体地,可以以36°角度、72°角度、108°角度、144°角度、180°角度间隔地沿所述搅拌轴4-7-1的周向设置;优选地,在以所述搅拌轴4-7-1为中心的垂直面上,多个所述刮板4-7-2以5-8cm间隔层叠地沿着所述搅拌轴4-7-1的轴向设置,优选为5-6cm间隔;所述脱单体反应器iv内部带有所述搅拌轴4-7-1,在脱单体过程中,通过旋转作用促使新的物料覆盖表面旧物料,使聚合物主体内部的单体在有限时间内停留,防止过热、避免发生再聚合。
另外,优选地,所述刮板4-7-2的外缘与所述脱单体反应器iv的内壁的间距为1-3cm,优选为1-2cm。在本发明中,聚乳酸高黏度熔体通过刮板,在内壁上形成均匀、超薄的膜状结构,该膜状结构可提供足够大的表面积,从而使残留丙交酯单体充分挥发出来。另外,在本发明中,需要说明的是,每一个所述刮板4-7-2具有两个长边和两个短边,其中的一个短边与所述搅拌轴4-7-1相接触,另一个短边的边缘即为所述刮板4-7-2的“外缘”。
根据本发明,所述脱单体反应器iv还设置有聚乳酸熔体入口4-4、导热油入口4-5、导热油出口4-6、丙交酯单体出口4-8和聚乳酸成品出口4-9。
根据本发明,所述第二聚合反应器iii的内部设置有搅动组件120。
优选地,所述搅动组件120为由带螺旋槽的鱼雷体、线圈架和电磁绕组组成的一体结构。
根据本发明,图3是本发明第二聚合反应器iii的示意图,如图3所示,所述第二聚合反应器iii还设置有静态混合器132以及与所述静态混合器132相连接设置的循环泵133,该静态混合器132的设置能够提高聚合熔体与助剂混合效率;在本发明中,所述第二聚合反应器iii还设置有熔体进料口111、熔体出料口112、换热液入口141和换热液出口142。
根据本发明,所述第一聚合反应器ii的内部设置有搅拌装置,以及在所述第一聚合反应器ii的外部设置有夹套。
根据本发明,所述装置还包括与所述脱单体反应器iv相连接设置的双螺杆挤出机v。
本发明还提供了一种由丙交酯制备聚乳酸的方法,其中,该方法在制备聚乳酸的装置中进行,包括以下步骤:
(a)将丙交酯在丙交酯熔融罐i中进行熔融处理,得到熔融态丙交酯;
(b)在催化剂和引发剂的存在下,将所述熔融态丙交酯和复合稳定剂在第一聚合反应器ii进行第一聚合反应,得到第一熔体;
(c)将所述第一熔体通入第二聚合反应器iii中进行第二聚合反应,得到第二熔体;
(d)在搅拌条件和激振力的共同作用下,将所述第二熔体通入脱单体反应器iv中进行脱单体反应;
其中,所述装置为前述所述的装置。
根据本发明,图5是本发明制备聚乳酸的装置的示意图,如图5所示,所述的制备聚乳酸的装置包括依次连接设置的丙交酯熔融罐i、第一聚合反应器ii、第二聚合反应器iii和脱单体反应器iv。
根据本发明,在步骤(a)中,所述熔融处理的条件包括:温度为90-100℃,时间为1-1.2h;优选地,温度为90-95℃,时间为1-1.1h。在本发明中,优选情况下,在连续地氮气的保护下,将丙交酯加入到丙交酯熔融罐i;其中,丙交酯可以为外消旋丙交酯,内消丙交酯、左旋丙交酯或右旋丙交酯,在本发明中,丙交酯购自totalcorbionpla(thailand)ltd,商品名为lumilact®l,净重600kg,型号为l85,纯度为99.5%。
根据本发明,在步骤(b)中,所述催化剂为辛酸亚锡。在本发明中,选择本发明所限定的催化剂的优点是当单体与催化剂比例较高的情况下,具有转化率高、消旋化现象少,聚合物分子量高的特点。
根据本发明,所述引发剂为聚乙二醇和/或异辛醇,优选为聚乙二醇。在本发明中,优选情况下,选择本发明所限定的引发剂聚乙二醇的优点是丙交酯的聚合从聚乙二醇的端羟基开始反应,生产聚乙二醇-聚乳酸的两嵌段聚合物,聚乙二醇在聚合过程中产生较短的分子链,低的引发剂含量产生高的聚合物分子量,得到较高的聚合度。
根据本发明,在第一聚合反应中,本发明的发明人发现,在聚乳酸聚合过程中,由于pla聚合过程中不可避免要受到热、氧、机械剪切等因素的影响,会发生热降解,使熔融黏度降低,引起最终制品物理性能降低。同时在聚合过程中也会产生氧化降解,氧化降解是一个具有链引发、链增长、链终止过程的自动氧化连锁反应,清除自由基和分解氢过氧化物是抑制聚合物氧化降解的基本途径。进一步,本发明的发明人发现,由于丙交酯存在热老化和热氧老化等副反应,导致得到的聚乳酸切片呈微黄色;通过添加复合稳定剂,能够将氢过氧化物分解成不活泼产物,抑止自动催化氧化过程,进一步地,在聚乳酸聚合过程中加入1-3‰的所述复合稳定剂,优选为2‰的所述复合稳定剂。能够很好的改善所制备的聚乳酸切片的色泽,尤其是能够降低切片色泽中的b值,比现有常规聚乳酸切片降低78.0%。
在本发明中,在所述复合稳定剂的选择上,本发明的发明人发现,目前广泛采用的抗氧剂主要是受阻酚和亚磷酸酯两大类。在抗氧过程中,受阻酚捕捉聚合物过氧化自由基(roo·)后变成氢过氧化物(rooh),氢过氧化物对氧化降解具有自动催化作用,而受阻酚自身不能分解氢过氧化物,所以单独使用时难以达到理想的抗氧效果。亚磷酸酯的抗氧作用发生在磷原子上,它能够分解氢过氧化物,由三价磷变成稳定的五价磷,但由于它不具有捕捉自由基的能力,单独使用也不能够令人满意。而将两者混合使用,作为复配稳定剂,使其作用相互补充,由于组分之间的协同作用,使抗氧和其它多方面效能得到了最大发挥,其性能优于任何单一组分的抗氧剂。
在抗氧剂筛选上从加工稳定性、长效稳定性及色泽稳定性三个方面考虑,选择主抗氧剂和辅助抗氧剂的复配。
其中,所述主抗氧剂为受阻酚类抗氧剂,所述受阻酚类抗氧剂为四[β-(3,5-二叔丁基-4-羟基苯基)丙酸]季戊四(醇)酯(at-10)和/或β-(3,5-二叔丁基-4-羟基苯基)丙酸十八酯(at-76)。
其中,所述辅助抗氧剂为亚磷酸酯类抗氧剂,所述亚磷酸酯类抗氧剂为三(2,4-二叔丁基苯基)亚磷酸酯(at-168)和/或双(2,4-二叔丁基苯基)季戊四醇二亚磷酸酯(at-626)。
优选情况下,所述受阻酚类抗氧剂和所述亚磷酸酯类抗氧剂的重量比为1:(0.1-3),更优选为1:(0.5-2),更进一步优选为1:(1-2)进行复配。在本发明中,将所述受阻酚类抗氧剂和所述亚磷酸酯类抗氧剂按照前述所限定的范围进行复配,能够更好地改善所制备的聚乳酸切片的色泽,尤其是能够更好地降低切片色泽中的b值。
根据本发明,最优选情况下,所述受阻酚类抗氧剂和所述亚磷酸酯类抗氧剂具体复配如下:
(1)复合稳定剂是由at-10与at-168按质量比1:1复配,标记为cs-1;
(2)复合稳定剂是由at-10与at-626按质量比1:1复配,标记为cs-2;
(3)复合稳定剂是由at-76与at-168按质量比1:1复配,标记为cs-3;
(4)复合稳定剂是由at-76与at-626按质量比1:1复配,标记为cs-4;
(5)复合稳定剂是由at-10与at-168按质量比1:2复配,标记为cs-5;
(6)复合稳定剂是由at-10与at-626按质量比1:2复配,标记为cs-6;
(7)复合稳定剂是由at-76与at-168按质量比1:2复配,标记为cs-7;
(8)复合稳定剂是由at-76与at-626按质量比1:2复配,标记为cs-8;
(9)复合稳定剂是由at-10与at-168按质量比2:1复配,标记为cs-9;
(10)复合稳定剂是由at-10与at-626按质量比2:1复配,标记为cs-10;
(11)复合稳定剂是由at-76与at-168按质量比2:1复配,标记为cs-11;
(12)复合稳定剂是由at-76与at-626按质量比2:1复配,标记为cs-12。
在本发明中,将所述受阻酚类抗氧剂和所述亚磷酸酯类抗氧剂按照如上限定的进行复配,能够很好的改善聚乳酸切片的色泽,其色值中的b值最低为4.19,比现有常规聚乳酸切片降低78.0%。
根据本发明,丙交酯、所述催化剂、所述复合稳定剂和所述引发剂的用量的重量比为1:(0.002-0.005):(0.001-0.002):(0.008-0.01);优选为1:(0.002-0.004):(0.001-0.015):(0.008-0.009)。在本发明中,更进一步优选地,将丙交酯、所述催化剂、所述复合稳定剂和所述引发剂的用量的重量比1:0.002:0.0015:0.008,能够得到成本与性能兼优的聚乳酸熔体。
根据本发明,在步骤(b)中,所述第一聚合反应的条件包括:温度为150-160℃,压力为50-53kpa,反应时间为3-3.2h;优选地,温度为155-160℃,压力为50-52kpa,反应时间为3-3.1h。
在本发明中,经所述第一聚合反应后,所得到的第一熔体的转化率高达50-55%。
根据本发明,在步骤(c)中,所述第二聚合反应的条件包括:温度为190-200℃,压力为6-6.5mpa,反应时间为1-1.2h;优选地,温度为195-200℃,压力为6-6.2mpa,反应时间为1-1.1h。
在本发明中,经所述第二聚合反应后,所得到的第二熔体的转化率高达95-97%。
另外,需要说明的是,在所述第二聚合反应中,由于熔体脉动形变装置和静态混合器相互作用,使得转化率得到提高。
根据本发明,在步骤(d)中,所述脱单体反应的条件包括:将所述第二次聚合反应后的熔体加入终止剂进行终止反应,其中,所述终止剂为亚磷酸和/或2-乙基辛酸;优选情况下,丙交酯与所述终止剂的用量质量比为1:(0.002-0.005),优选为1:(0.002-0.003)。
温度为210-215℃,压力为1-1.2kpa,反应时间为0.5-0.6h;搅拌速率为10-30rpm;振幅为0.15-0.3mm,振频为20-45hz。在本发明中,在所述脱单体反应过程中,所述脱单体反应器iv中的所述电磁激振器4-1与通过振动传递盘与搅拌驱动电机4-2相连接以使所述搅拌驱动电机4-2进行轴向振动,能够有效地降低第二熔体中残留的丙交酯的含量,进而能够使得到的聚乳酸切片丙交酯含量较好地降低。另外,所述搅拌轴4-7-1随所述电磁激振器4-1激振力的作用进行轴向振动,所述搅拌轴4-7-1上的所述刮板4-7-2也随着所述搅拌轴4-7-1振动发生周期性变化,将振动力场引入聚乳酸熔体,聚乳酸高黏度熔体通过该振动刮板,在内壁上形成均匀、超薄的膜状结构,该膜状结构可提供足够大的表面积,在生产过程中,使用所述脱单体反应器iv能够实现较大成膜面积和较快表面更新的结合,有助于提高熔体的分散混合,提高丙交酯单体挥发量,满足快速脱除单体的要求,能够将残留聚乳酸熔体的丙交酯单体含量控制在最低程度。
根据本发明,该方法还包括将经步骤(d)后得到的聚乳酸熔体进行水冷切粒、脱水、结晶和干燥处理;在本发明中,将经步骤(d)后的聚乳酸熔体经熔体管道以及送(出)料泵通入双螺杆挤出机v中,经水冷切粒、脱水、筛选、结晶、除湿干燥,最终得到聚乳酸树脂,其中,造粒机温度为205-210℃。
其中,水冷切粒转速1500-3000rpm,脱水转速800-1000rpm,结晶温度为90-95℃、时间为35-45min,除湿干燥温度为90-98℃、时间为1-2h;优选地,结晶温度为95℃、时间为40min,除湿干燥温度为95℃、时间为1.5h。
本发明还提供了一种前述所述的方法制备得到的聚乳酸。
根据本发明,所述聚乳酸的重均分子量为8万-25万,所述聚乳酸中丙交酯单体的含量为0.8-1.0wt‰,所述聚乳酸在2.16kg载荷下190℃的熔融指数6-27g/10min;优选地,所述聚乳酸的重均分子量为10万-25万,所述聚乳酸中丙交酯单体的含量为0.8-0.9‰,所述聚乳酸在2.16kg载荷下190℃的熔融指数8-27g/10min。
通过上述技术方案,通过采用本发明的装置和方法制备聚乳酸,尤其是在丙交酯聚合过程中加入复合稳定剂,能够很好的改善聚乳酸切片的色泽缺陷,最终产品色值中的b值为4.19,比现有常规聚乳酸切片降低78%;脱单体反应过程中,脱单体器旋转轴采用电磁激振器进行轴向振动,能够有效地降低聚乳酸熔体残留丙交酯单体含量,最终得到的聚乳酸切片丙交酯单体含量为0.8‰,比现有常规工艺生产的pla切片降低80.5%。另外,采用该装置和方法制备的聚乳酸切片黄度低,脱挥效率高,实现了高品质pla切片的产业化生产。
本发明获得的聚乳酸(溶体)可以进一步制备成改性聚乳酸材料,因此,本发明提供了一种熔体在线制备改性聚乳酸材料的装置,其中,所述装置包括双螺杆挤出机、第一固体改性剂料斗8c、聚乳酸熔体管道14c和聚乳酸熔体进料计量泵10c,且所述第一固体改性剂料斗8c与所述双螺杆挤出机的一区2c相连接,所述聚乳酸熔体管道14c与所述双螺杆挤出机的四区4c相连接。
在本发明中,需要说明的是,聚乳酸熔体由中粮生化自制,制备方法包括:将丙交酯经聚合和脱挥工序得到聚乳酸熔体;其中,该丙交酯购自totalcorbionpla(thailand)ltd,商品名为lumilact®l,净重600kg,型号为l85,纯度为99.5%。
根据本发明,所述装置还包括第二固体改性剂料斗9c和第二辅料料斗13c,其中,所述第二固体改性剂料斗9c与所述双螺杆挤出机的一区2c相连接;所述第二辅料料斗13c与所述双螺杆挤出机的五区5c相连接。
根据本发明,所述装置还包括第一辅料料斗11c,所述第一辅料料斗11c与所述双螺杆挤出机的一区2c相连接;用于粉剂辅料的进料。
根据本发明,所述双螺杆挤出机还包括液体进料计量泵12c,且所述液体进料计量泵12c与所述双螺杆挤出机的二区3c相连接。
根据本发明,如图8本发明的在线制备改性聚乳酸材料的装置的示意图所示,所述双螺杆挤出机还包括电机1c和双螺杆挤出机机头7c;另外,双螺杆挤出机一区2c、双螺杆挤出机二至三区3c、双螺杆挤出机四区4c、双螺杆挤出机五区5c、双螺杆挤出机六至十一区6c如图8标识所示。
本发明还提供了一种在线制备改性聚乳酸材料的方法,其中,该方法在在线制备改性聚乳酸材料的装置中进行,该方法包括:
(a)将聚乳酸熔体经聚乳酸熔体管道14c进入双螺杆挤出机的四区4c;
(b)将第一固体改性剂经第一固体改性剂料斗8c进入所述双螺杆挤出机的一区2c;
(c)将液体改性剂经液体进料计量泵12c进入所述双螺杆挤出机的二区3c;
(d)所述聚乳酸熔体、所述第一固体改性剂和所述液体改性剂在所述双螺杆挤出机的五区5c至十一区6c进行接触,并将得到改性聚乳酸聚合熔体挤出造粒;
其中,所述在线制备改性聚乳酸材料的装置为前述所述的装置。
根据本发明,所述液体改性剂选自环氧大豆油、柠檬酸三丁酯和己二酸二乙二醇单丁醚酯中的一种或多种。
其中,环氧大豆油在常温下为浅黄色黏稠油状液体,分子式为c57h106o10,分子量为950,环氧值≥6.60,碘价≤3.0,环氧值eso越高,耐热性越好,碘价越低和pvc的兼容性越好,越不容易析出。
其中,柠檬酸三丁酯,通常称柠檬酸三正丁酯,化学名3-羟基-3-羧基戊二酸三丁酯,是一种酯类化合物。
根据本发明,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为49.5-79重量%,所述第一固体改性剂的用量为20-50重量%,所述液体改性剂的用量为0.2-0.5重量%;优选地,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为53-66.8重量%,所述第一固体改性剂的用量为25-35重量%,所述液体改性剂的用量为0.2-0.3重量%。在本发明中,所述聚乳酸熔体、所述第一固体改性剂和所述液体改性剂的用量的总和为百分之百。
根据本发明,该方法还包括:在步骤(b)中,将第一固体改性剂经第一固体改性剂料斗8c以及将第二固体改性剂经第二固体改性剂料斗9c进入所述双螺杆挤出机的一区2c。
根据本发明,所述第一固体改性剂和所述第二固体改性剂相同或不同,各自为增韧剂和/或增强剂。
在本发明中,所述增韧剂选自聚己二酸对苯二甲酸丁二醇酯、聚丙撑碳酸酯、聚丁二酸丁二醇酯、聚丁二醇-丁二酸/己二酸共聚酯中的一种或多种,优选为聚己二酸对苯二甲酸丁二醇酯和/或聚丙撑碳酸酯。
在本发明中,所述增强剂为滑石粉和/或碳酸钙。
根据本发明,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为49.5-79重量%,所述第一固体改性剂的用量为20-50重量%,所述第二固体改性剂的用量为0-7重量%,所述液体改性剂的用量为0.2-0.5重量%;优选地,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为53-66.8重量%,所述第一固体改性剂的用量为25-35重量%,所述第二固体改性剂的用量为3-7重量%,所述液体改性剂的用量为0.2-0.3重量%。在本发明中,所述聚乳酸熔体、所述第一固体改性剂、所述第二固体改性剂和所述液体改性剂的用量的总和为百分之百。
根据本发明,该方法还包括:在步骤(d)中,将所述聚乳酸熔体、所述第一固体改性剂、所述液体改性剂和辅料进行接触,其中,所述辅料经所述第二辅料料斗13c进入所述双螺杆挤出机的五区5c。
根据本发明,所述辅料选自扩链剂、交联剂、抗氧剂和润滑剂中的一种或多种。
根据本发明,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为49.5-79重量%,所述第一固体改性剂的用量为20-50重量%,所述液体改性剂的用量为0.2-0.5重量%,所述扩链剂的用量为0-0.3重量%,所述交联剂的用量为0-0.3重量%,所述抗氧剂的用量为0-0.5重量%、所述润滑剂的用量为0-0.4重量%;优选地,优选地,以所述改性聚乳酸熔体的总重量为基准,所述聚乳酸熔体的用量为53-66.8重量%,所述第一固体改性剂的用量为25-35重量%,所述液体改性剂的用量为0.2-0.3重量%,所述扩链剂的用量为0.1-0.2重量%,所述交联剂的用量为0.1-0.2重量%,所述抗氧剂的用量为0.2-0.3重量%,所述润滑剂的用量为0.2-0.3重量%。在本发明中,所述聚乳酸熔体、所述第一固体改性剂、所述液体改性剂、所述扩链剂、所述交联剂、所述抗氧剂和所述润滑剂的用量的总和为百分之百。
本发明还提供了一种由前述所述的方法制备得到的改性聚乳酸材料。
通过上述技术方案,本发明的有益效果是,将聚合得到的聚乳酸熔体通过在线添加改性剂实现在线改性,省去了二次加工,一方面大大降低了生产成本,另一方面可以避免聚乳酸因二次加工发生的降解,保证了最终制品的优异性能。
本发明提供了一种聚乳酸脱挥蒸发器,其中,所述聚乳酸脱挥蒸发器包括:
容器,所述容器包括竖直延伸的圆管形的筒体3;
搅拌轴5,所述搅拌轴5至少部分地设置在所述筒体3中,并且所述搅拌轴5与所述筒体3共轴;
搅拌带6,所述搅拌带6连接于所述搅拌轴5,所述搅拌带6设置为围绕所述筒体3的中心轴线的螺旋状,所述搅拌带6包括朝向所述筒体3的内周面的外侧带面,并且所述外侧带面与所述筒体3的内周面彼此间隔。在本发明中,在未作相反说明的情况下,使用的方位词如“上、下”通常是指容器的筒体竖直延伸设置时的位置关系。
本方案的聚乳酸脱挥蒸发器可以用于聚乳酸的脱挥工艺,当然也可以应用于具有类似性质的聚合物的脱挥工艺。其中,所述容器容纳待处理物料和所述蒸发器的内构件,其包括筒体3,筒体3形成为圆管形,在使用时可以竖直延伸设置,便于使用转动的搅拌件对物料进行搅拌处理;搅拌轴5为传动部件,其可以连接于动力驱动件,例如发动机、电机等,以被驱动为围绕筒体3的中心轴线旋转;搅拌带6连接于搅拌轴5,以跟随搅拌轴5转动。另外,所述聚乳酸脱挥蒸发器包括穿过所述筒体3的进料管1以及连接于所述进料管1的分布器2,所述分布器2包括围绕所述筒体3的中心轴线周向延伸的环状管,所述环状管上设置有布料口15。如图9所示,进料管1穿过筒体3并连接于筒体3中的分布器2,分布器2为周向的环状管,并且其上形成有布料口15,进料管1可以将物料输送到分布器2中,并通过布料口15将物料较为均匀地分布到筒体3的内壁上。在其它实施方式中,也可以使用从容器的其他部分(如密封盖13)伸入筒体3内部的进料管,可以使用或不使用分布器2。
进一步的,所述分布器2位于所述搅拌带6的上侧,所述布料口15朝向所述筒体3的内周面,并且所述布料口15的朝向优选向下倾斜。分布器2上的布料口15可以形成在径向外侧,以朝向筒体3的内周面,使得物料喷出后可以粘附在筒体3的内周面上并在重力作用下沿着筒体3的内壁向下流动。优选的方案是这些布料口15稍微向下倾斜,从而使得物料从布料口喷出后到达筒体3的内周面时可以更靠近下方的搅拌带6的高度位置。布料口15可以为孔、狭缝等。
其中,参考图9所示,搅拌带6形成为围绕筒体3的中心轴线的螺旋状,具有朝向筒体3的内周面的外侧带面,并且该外侧带面与筒体3的内周面保持间隔。螺旋状搅拌带6的螺距,可以为筒体3内径的0.1-2倍,优选0.2-1.2倍,最优选0.3-0.8倍;螺旋状搅拌带6外侧带面的宽度,可以为筒体3内径的0.01-0.15倍,优选0.02-0.1倍,最优选0.03-0.08倍。当筒体3的内周面上粘附有粘性物料时,转动的搅拌带3可以使得物料位于外侧带面和筒体3内周面之间,另外,根据搅拌带6的螺旋方向(指左旋或右旋),通过调节搅拌轴5的转动方向(指顺时针或逆时针),使得搅拌带6可以将筒体3内周面上的物料向下刮,使得物料更为均匀地涂在筒体3的内周面上。物料在筒体3的边壁上受热后,随着其中含有的小分子挥发性组分逐渐蒸发,其黏度也逐渐升高。通过搅拌带6施加在物料上的刮擦力,可以使已脱除了部分挥发分呈粘稠浆体状的物料贴着筒体内壁螺旋式地向下移动,这样既可以防止物料因黏度过大黏附在筒体内壁上固定不动,又可以防止物料在筒体内壁局部区域积聚成坨后直接落入筒体底部而无法得到充分加热,以达到使物料在脱挥蒸发器内能够得到相对均匀地加热的目的,从而防止物料因过度加热而导致的颜色变深和热分解,同时也避免部分物料因得不到充分加热而导致的脱挥不充分问题,以提高脱挥效果。
关于搅拌带6的转动方向,参考图9,其中搅拌带6为左旋,搅拌带6顺时针转动,即可通过各个位置的下侧边缘将物料轴向向下刮,当搅拌带为右旋时,相应的采用逆时针转动即可。搅拌带6的核心结构在于外侧带面,搅拌带6并不局限为带状结构,只要其具有与筒体3内周面间隔的外侧带面即属于本方案的保护范围,当然,形成为带状为一种优选的实施方式。
可选择的,所述外侧带面与所述内周面的间隔为2mm-30mm。搅拌带6在不同位置的外侧带面与内周面的间隔可以保持一致,从而在筒体3的内周面上形成均匀的物料层,该间隔可以为2mm-30mm,从而可以在筒体3的内周面上形成2mm-30mm的物料层。
在图9所示的实施方式中,所述搅拌带6与所述搅拌轴5径向间隔,并且所述搅拌带6通过连接杆19连接于所述搅拌轴5。参考图9所示,搅拌轴5可以从筒体3的上部延伸到下部,其外径小于搅拌带6所形成的螺旋结构的内径,并且搅拌带6通过连接杆19连接于搅拌轴5,因此搅拌带6和搅拌轴5之间形成较大的空间,允许物料脱挥时产生的气态流体排出。当然,所述搅拌带6和所述搅拌轴5还可以通过其它的方式进行连接,例如,可以在搅拌带6和搅拌轴5之间设置一个开孔螺旋板,将螺旋板的外沿与螺带6焊接,内沿与搅拌轴5进行焊接,从而将两者连接一个刚性体,使搅拌轴可以带动搅拌带在蒸发器内转动,而蒸发器内蒸发出来的挥发性气体,可以通过螺旋板上的开孔上升到蒸发器的上部空间,再经排放口8排放出去。
进一步的,所述聚乳酸脱挥蒸发器包括设置在所述容器底部的支撑架12,所述搅拌轴5的下端可转动地支撑在所述支撑架12上。参考图9所示,支撑架12大致形成为三角架的形式,以支撑搅拌轴5的下端,并允许搅拌轴5转动。支撑架12可以支撑在锥形筒9的锥形内周面上,当然,在其它实施方式中,支撑架12也可以支撑在筒体5的内周面上。
如图9所示,所述聚乳酸脱挥蒸发器包括包裹在所述筒体3外部的夹套4,所述夹套4和所述筒体3之间形成密封腔。所述夹套4的上端设置有蒸汽入口管14,下端设有蒸汽凝液出口管7。通入夹套4的蒸汽通过筒体3将热量传递给沿着筒体内壁向下蠕动的粘性物料,从而使物料中沸点相对较低的组份如稀释催化剂的溶剂、未聚合的丙交酯单体或聚合过程因为水解等副反应产生的小分子酸或酯类物质气化成气态从物料中挥发出来。当然,也可以使用导热油或热水作为加热介质。但采用液体加热介质时,应该使加热介质从夹套4的底部进入,从夹套4的顶部排出。为了减少系统对环境散热导致的热损失并防止烫伤事故,一般需在夹套4外设置厚度不小于5cm的隔热保温层。
另外,所述容器包括连接于所述筒体3上端的密封盖13和设置在所述密封盖上的密封函18,所述搅拌轴5可转动地穿过所述密封盖13和所述密封函18,所述搅拌轴5和所述密封函18之间设置有密封填料17,并且优选在所述密封函18上形成有密封气体注入管16。参考图9所示,密封盖13密封在筒体3的上端,并且密封盖13一体连接有密封函18,密封函18大致形成为管状,搅拌轴5穿过密封盖13和密封函18,且通过密封填料17环绕搅拌轴5,提高密封性能。另外,优选在密封函18的侧部设置密封气体注入管16,通过密封气体注入管16可以向密封函18和搅拌轴5之间的缝隙注入气体,使得该缝隙内能够维持微正压,可以避免外部气体或杂质进入到密封函18和搅拌轴5之间。注入的气体可以为惰性气体,如氮气、氩气等。
此外,所述筒体3的上部设置有能够连接于真空设备的排放管8,所述筒体3的下端连接有锥形筒9,所述锥形筒9的下端形成出料口10,所述锥形筒9的侧壁连接有惰性气体注入管11。锥形筒9的内径向下不断缩小,可以通过底部的出料口10排出脱挥后的物料;通过惰性气体注入管11可以向筒体3中注入保护气体,排出内部的空气,以避免其中的物料与空气发生化学反应;排放管8可以连接于真空设备,以对筒体3抽真空,降低其中的压力,便于物料脱挥蒸发,另外,通过惰性气体注入管11注入气体,也可以降低脱挥生成的气态物的分压,促进脱挥作用。
根据本方案的另一种实施方式,如图10所示,所述聚乳酸脱挥蒸发器围绕所述筒体3的中心轴线周向间隔的多个支撑杆20、连接于所述支撑杆20两端的上连接座21和下连接座25,其中上连接座21连接于所述搅拌轴5,所述搅拌轴5为中空的管状结构,在所述管状搅拌轴5的侧壁上沿周向开设了4-16个排气孔27,这些开孔的总面积与管状搅拌轴的横截面积大致相当。在上连接座21的中心设置了排气孔26,该孔的截面积也和管状搅拌轴的横截面积大致相当。物料在蒸发器内受热后蒸发出来的挥发性气态组份,可以通过上连接座25的排气孔进入搅拌轴5的内腔,再从搅拌轴5侧壁上的排气孔排入蒸发器上部的空间,最后从排放口8排出蒸发器。所述多个支撑杆贴合连接于所述搅拌带6的径向内侧表面。所述搅拌带6的径向外侧表面和筒体3的内侧表面之间的间隙为2-30mm。参考图10所示,多个支撑杆20和两个连接座21组成搅拌带6的支撑结构,支撑杆20贴合于搅拌带6的径向内侧表面,这同样在搅拌带6围成的内部空间形成允许气体排出的通道。上端的连接座21连接于搅拌轴5,以通过搅拌轴5带动支撑杆20和搅拌带6转动。上连接座21的下端外沿和筒体3内壁之间的缝隙宽度为1-25mm,从进料管1引入的物料进入蒸发器后,通过上连接座21和筒体3内壁之间的缝隙沿着筒体3的内壁往下流动。当进料管1的出口不设置分布器而利用上连接21和筒体3内壁之间的缝隙进行料液分布时,最好使进料管1沿筒体3的切向与筒体3连通。
在如图10所示的实施方式中,在筒体3外壁焊接了加热螺旋盘管22,导热油或高温热水通过加热介质入口管24进入加热螺旋盘管22,再从液态加热介质出口管23流出。为了减少系统对环境散热导致的热损失并防止烫伤事故,一般需在盘管外设置厚度不小于5cm的隔热保温层。
本发明提供的蒸发器用于聚乳酸脱挥时,通入夹套4或加热螺旋盘管22的加热介质的入口温度范围为180-240℃,优选的温度范围是190-230℃,最优选的温度范围是200-225℃;蒸发器内压力的范围为0.1kpa-20kpa,优选的压力范围是1kpa-10kpa,最优选的压力范围是2kpa-8kpa;搅拌轴的转速范围为5-120转/分钟,优选10-60转/分钟,最优选15-45转/分钟。
通过采用本发明提供的技术方案,物料进入脱挥装置后,首先能够相对均匀地分布到脱挥蒸发器上部的筒体内表面接受加热,从而使其中的易挥发组分气化进入筒体内部的气相空间。随着物料中含有的小分子挥发性组分不断蒸发,物料在沿着筒体边壁向下流动的过程中其黏度也逐渐升高。物料中挥发性组分含量降低到一定程度以后,因为粘度过高,单纯依靠自身重力已无法使物料继续顺畅地沿着边壁向下流动。本发明通过在蒸发器搅拌轴的周围贴近筒体边壁的位置设置螺旋形搅拌带,随着搅拌轴带动搅拌带在蒸发器内转动,可以对黏附在筒体内壁上的物料施加一个倾斜向下的刮擦力,从而可以使已脱除了部分挥发分呈粘稠浆糊状的物料能够贴着筒体内壁继续螺旋式地向下移动,这样既可以防止物料因黏度过大黏附在筒体内壁上,又可以防止物料在筒体内壁局部区域积聚成坨后在重力作用下直接落入筒体底部而无法得到充分加热,从而使物料在整个脱挥过程中都能够比较均匀地受热,并且在脱挥蒸发器内高温环境下的停留时间长度也大体相同,因而有利于提高脱挥后聚乳酸产品的质量。此外,在本发明提供的脱挥蒸发器中,需要脱除的挥发性组分受热气化后,能够迅速从筒体边壁的薄层物料中逸出,然后通过筒体内部的气态物料流通通道顺畅地从蒸发器内排出,因而可以获得较高的脱挥效率。
以下将通过实施例对本发明进行详细描述。以下实施例中,除非特别说明,所用试剂、培养基均为市售商品,所用方法均为常规方法。
实施例1.1-1.2和参比实施例1.1
这些实施例用来说明本发明涉及的乳酸发酵菌种。其中:
1、培养基
mrs液体培养基:蛋白胨10g、酵母浸粉5g、牛肉膏10g、葡萄糖20g、磷酸氢二钾2g、柠檬酸二铵2g、无水乙酸钠5g、硫酸锰0.25g、硫酸镁0.58g、吐温801ml、蒸馏水1000ml,ph6.5,121℃灭菌20min;
产酸发酵培养基:葡萄糖180g/l、酵母提取物10g/l、乙酸钠2g/l、kh2po40.5g/l、mgso4•7h2o0.5g/l、mnso40.2g/l、吐温801ml/l、caco390g/l。
2、乳酸含量通过高效液相色谱法进行检测:
色谱仪:agilenttechnologies1260infinityii;
检出器:rid;
分离柱:aminexhpx-87hcolumn300×7.8mm;
流动相:0.05m硫酸;
流量:0.5ml/min;
进样量:20μl;
乳酸保留时间为14min左右。
3、葡萄糖和l-乳酸的含量通过生物传感器进行检测:
仪器:sba-40e型生物传感器;
酶膜:d-葡萄糖酶膜和l-乳酸酶膜;
进样量:25μl。
4、乳酸的光学纯度通过高效液相色谱法进行检测:
色谱仪:agilenttechnologies1260infinity;
检出器:波长254nm,灵敏度0.32aufs;
分离柱:mcigel-crs10w(3u)4.6id×50mm;
流动相:0.002m硫酸铜;
流量:0.5ml/min;
进样量:20μl。
将样品稀释至总乳酸浓度为0.5-1g/l再进行检测。d-乳酸保留时间为11min左右,l-乳酸保留时间为13min左右,根据其峰面积计算l-乳酸的光学纯度。
糖酸转化率的计算公式为:发酵结束时刻的乳酸总质量/初始的葡萄糖质量。
实施例1.1
挑取鼠李糖乳杆菌菌株cgmccno.19507的单菌落接种于mrs液体培养基,37℃、150rpm过夜培养获得od600为12的种子液。然后,在200ml产酸发酵培养基中按照10%(v/v)的比例接种上述种子液,37℃、150rpm摇床培养6小时让菌株生长,然后升温到48℃,继续以150rpm摇床培养42小时获得发酵液(总发酵时长48小时)。发酵结束后通过高效液相色谱测定乳酸总产量和l-乳酸光学纯度。
其结果是,在200ml摇瓶水平下48℃发酵时,鼠李糖乳杆菌菌株cgmccno.19507发酵48h得到的发酵液中乳酸含量分别为216g/l,糖酸转化率为97%,l-乳酸光学纯度为99.8%。
实施例1.2
按照实施例1.1的方法发酵产乳酸,不同的是,将鼠李糖乳杆菌菌株cgmccno.19507替换为鼠李糖乳杆菌菌株cgmccno.19508,其结果是,在200ml摇瓶水平下48℃发酵时,鼠李糖乳杆菌菌株cgmccno.19508发酵48h得到的发酵液中乳酸含量分别为215.3g/l,糖酸转化率为96.5%,l-乳酸光学纯度为99.5%。
参比实施例1.1
按照实施例1.1的方法发酵产乳酸,不同的是,将鼠李糖乳杆菌菌株cgmccno.19507替换为鼠李糖乳杆菌菌株cgmccno.16834(该菌株已在cn109628339a中公开);
结果得到的发酵液中乳酸含量为157g/l,l-乳酸光学纯度为99%。
由实施例1.1-1.2和参比实施例1.1的结果可以看出,鼠李糖乳杆菌菌株cgmccno.19507和鼠李糖乳杆菌菌株cgmccno.19508在l-乳酸的产率和产物光学纯度方面优于鼠李糖乳杆菌cgmccno.16834,因此,本发明提供了一种发酵成本低、环境友好、l-乳酸生产速度快并且产物光学纯度高的新型生产菌株,为l-乳酸的工业化微生物发酵生产提供了更优的潜在选择。
实施例2.1-2.7与参比实施例2.1-2.2
这些实施例用来说明本发明涉及的发酵制备d-乳酸的方法。其中:
干物含量是指将测试样品在130℃左右干燥40min后的质量占干燥前的样品质量的百分数。
所使用的淀粉乳为:中粮生化能源(榆树)有限公司的淀粉乳。石灰乳中ca(oh)2的含量为20重量%。淀粉酶为:诺维信公司的耐高温α-淀粉酶、β-淀粉酶和异淀粉酶的复合酶体系,重量比为1:0.1:0.2。糖化酶为诺维信公司的amg300l。玉米浆为中粮生化能源(榆树)有限公司的浓玉米浆。纯葡萄糖为国药集团化学试剂有限公司的葡萄糖。
所使用的发酵罐为上海保兴生物工程设备有限公司的biotech-100js型号的100l发酵罐。
未作特殊说明的情况下,采用的发酵菌株为保藏编号为cgmccno.16835的植物乳杆菌(参见cn109504630a),-80℃超低温冰箱保存,甘油管每隔1-3个月活化复培一次。
d-乳酸的含量通过hplc方法进行测定,条件包括:aminexhpx-87hcolumn300×7.8mm色谱柱,柱温:55℃,流动相:0.005m硫酸,流量:0.5ml/min,进样量:20μl,示差折光检测器。
实施例2.1
淀粉糖制备:ph为5.8的30kg淀粉乳(其中淀粉干物含量为40重量%)中加入6g淀粉酶(相当于200u/千克淀粉干基),用蒸汽在110℃下进行喷射液化(时间约为4s),然后闪蒸(压力差-0.08±0.02mpa),并于95℃中保温60min。经热交换降温至60℃,获得液化液。调节ph至5,加入糖化酶,重量为淀粉干基的0.5‰(相当于1000u/千克淀粉干基),搅拌均匀,60℃保温24h,获得de值达到99%的糖化液。
配罐:将上述糖化液直接加入发酵罐,同时加入灭菌后的玉米浆,加入量使得发酵液中氮元素的含量为3g/l,控制发酵液中的葡萄糖含量为190g/l(使得发酵液中碳元素的含量为76g/l)。加入20%发酵液重量的石灰乳控制发酵体系ph约为6±0.2,无需监控ph,控制发酵温度为40±3℃,搅拌转速为100rpm。
发酵:将培养好的od600=10的种子液以10%发酵液体积的接种量接入发酵罐,培养至葡萄糖耗尽。
结果:发酵周期50h,总糖酸转化率99%,d-乳酸产量达188g/l。
实施例2.2
淀粉糖制备:ph为5.8的30kg淀粉乳(其中淀粉干物含量为40重量%)中加入6g淀粉酶(相当于200u/千克淀粉干基),用蒸汽在110℃下进行喷射液化(时间约为2s),然后闪蒸(压力差-0.08±0.02mpa),并于95℃中保温90min,然后经热交换降温至60℃,获得液化液。调节ph至5,加入糖化酶,重量为淀粉干基的0.4‰(相当于800u/千克淀粉干基),搅拌均匀,60℃保温24h,获得de值达到96%的糖化液。
配罐:将上述糖化液直接加入发酵罐,同时加入灭菌后的玉米浆,加入量使得发酵液中氮元素的含量为3g/l,控制发酵液中的葡萄糖含量为190g/l(使得发酵液中碳元素的含量为76g/l)。加入25%发酵液重量的石灰乳控制ph约为6.2±0.2,无需监控ph,控制发酵温度为40±3℃,搅拌转速为100rpm。
发酵:将培养好的od600=10的种子液以10%发酵液体积的接种量接入发酵罐,培养至葡萄糖耗尽。
结果:发酵周期48h,糖酸转化率95%,d-乳酸产量达180g/l。
实施例2.3
淀粉糖制备:ph为5.8的30kg淀粉乳(其中淀粉干物含量为30重量%)中加入6g淀粉酶(相当于200u/千克淀粉干基),用蒸汽在110℃下进行喷射液化(时间约为3s),然后闪蒸(压力差-0.08±0.02mpa),并于95℃中保温60min。经热交换降温至60℃,获得液化液。调节ph至5,加入糖化酶,重量为淀粉干基的0.5‰(相当于1000u/千克淀粉干基),搅拌均匀,60℃保温24h,获得de值达到99%的糖化液。
配罐:将上述糖化液直接加入发酵罐,同时加入灭菌后的玉米浆,加入量使得发酵液中氮元素的含量为3g/l,控制发酵液中的葡萄糖含量为160g/l(相当于使得发酵液中碳元素的含量为64g/l)。加入25%发酵液重量的石灰乳控制ph约为6.2±0.2,无需监控ph,控制发酵温度为40±3℃,搅拌转速为100rpm。
发酵:将培养好的od600=10(相当于3.33g细胞干重/l)的种子液以10%发酵液体积的接种量接入发酵罐,培养至葡萄糖耗尽。
结果:发酵周期44h,糖酸转化率99%,d-乳酸产量达158g/l。
实施例2.4
淀粉糖制备:ph为5.8的30kg淀粉乳(其中淀粉干物含量为40重量%)中加入3g淀粉酶(相当于100u/千克淀粉干基),用蒸汽在100℃下进行喷射液化(时间约为1s),然后闪蒸(压力差-0.08±0.02mpa),并于80℃中保温90min,然后经热交换降温至60℃,获得液化液。调节ph至5,加入糖化酶,重量为淀粉干基的0.5‰(相当于1000u/千克淀粉干基),搅拌均匀,55℃保温20h,获得de值达到95%的糖化液。
配罐:将上述糖化液直接加入发酵罐,同时加入灭菌后的玉米浆,加入量使得发酵液中氮元素的含量为3g/l,控制发酵液中的葡萄糖含量为190g/l(相当于使得发酵液中碳元素的含量为76g/l)。加入18%发酵液重量的石灰乳控制ph约为5.8±0.5,无需监控ph,控制发酵温度为38±1℃,搅拌转速为50rpm。
发酵:将培养好的od600=10的种子液以10%发酵液体积的接种量接入发酵罐,培养至葡萄糖耗尽。
结果:发酵周期52h,糖酸转化率95%,d-乳酸产量达180g/l。
实施例2.5
淀粉糖制备:ph为5.8的30kg淀粉乳(其中淀粉干物含量为40重量%)中加入6g淀粉酶(相当于200u/千克淀粉干基),用蒸汽在120℃下进行喷射液化(时间约为4s),然后闪蒸(压力差-0.08±0.02mpa),并于100℃中保温90min,然后经热交换降温至65℃,获得液化液。调节ph至3,加入糖化酶,重量为淀粉干基的1‰(相当于2000u/千克淀粉干基),搅拌均匀,65℃保温20h,获得de值达到98%的糖化液。
配罐:将上述糖化液直接加入发酵罐,同时加入灭菌后的玉米浆,加入量使得发酵液中氮元素的含量为3g/l,控制发酵液中的葡萄糖含量为160g/l(相当于使得发酵液中碳元素的含量为64g/l)。加入25%发酵液重量的石灰乳控制ph约为6.2±0.3,无需监控ph,控制发酵温度为44±1℃,搅拌转速为150rpm。
发酵:将培养好的od600=10的种子液以10%发酵液体积的接种量接入发酵罐,培养至葡萄糖耗尽。
结果:发酵周期47h,糖酸转化率96%,d-乳酸产量达154g/l。
实施例2.6
采用实施例2.1的方法,不同的是,采用柠檬酸钠作为发酵过程中的中和剂,加入量和加入方式为:向发酵体系中持续流加10重量%的柠檬酸钠溶液,以使发酵体系ph控制在6。
结果:发酵周期48h,糖酸转化率90%,d-乳酸产量达171g/l。
实施例2.7
采用实施例2.1的方法,不同的是,采用氢氧化钠为发酵过程中的中和剂,加入量和加入方式为:向发酵体系中持续流加4重量%的氢氧化钠溶液,以使发酵体系ph控制在6±0.2。
结果:发酵周期38h,糖酸转化率100%,d-乳酸产量达160g/l。
实施例2.8
采用实施例2.1的方法,不同的是,采用下述方法进行糖化液制备:
ph为5.8的11.25kg淀粉乳(其中淀粉干物含量为15重量%)中加入2.25g淀粉酶(相当于200u/千克淀粉干基),用蒸汽在120℃下进行喷射液化(时间约为4s),然后闪蒸(压力差-0.08±0.02mpa),并于100℃中保温90min,然后经热交换降温至65℃,获得液化液。调节ph至3,加入糖化酶,重量为淀粉干基的1‰(相当于2000u/千克淀粉干基),搅拌均匀,65℃保温20h,获得de值达到97%的糖化液。
结果:发酵周期45h,总糖酸转化率96%,d-乳酸产量达155g/l。
参比实施例2.1
采用实施例2.1的方法,不同的是,采用纯葡萄糖液替换淀粉糖作为碳源。
结果:发酵周期44h,糖酸转化率100%,d-乳酸产量达190g/l。
参比实施例2.2
采用实施例2.1的方法,不同的是,采用鼠李糖杆菌(cgmccno.16834,公开于cn109628339a)作为发酵菌种。
结果:发酵周期80h,糖酸转化率12%,d-乳酸产量达19g/l。
参比实施例2.3
采用实施例2.1的方法,不同的是,控制搅拌转速为180rpm,加向发酵体系中持续流加4重量%的氢氧化钠溶液,以使发酵体系ph控制在5.5±0.2。
结果:发酵周期63h,总糖酸转化率83%,d-乳酸产量达133g/l。
参比实施例2.4
采用实施例2.1的方法,不同的是,将培养好的od600=10的种子液以20%发酵液体积的接种量接入发酵罐,控制搅拌转速为180rpm。
结果:发酵周期39h,总糖酸转化率88%,d-乳酸产量达141g/l。
参比实施例2.5
采用实施例2.1的方法,不同的是,所述糖化液的加入量使得发酵液中碳元素的含量为50g/l,并加向发酵体系中持续流加4重量%的氢氧化钠溶液,以使发酵体系ph控制在5±0.2。
结果:发酵周期38h,总糖酸转化率93%,d-乳酸产量达116g/l。
通过实施例2.1和参比实施例2.1的结果对比可看出,本发明提供的方法采用糖化液(淀粉糖)作为乳酸发酵的主要碳源,其发酵周期、糖酸转化率和d-乳酸产量和传统采用葡萄糖作为碳源的发酵工艺差别不大。但是本发明提供的方法在原料成本上较传统工艺更为低廉。以实施例2.1和参比实施例2.1中的糖化液和葡萄糖用量为例,实施例2.1中的淀粉原料成本约为60元,参比实施例2.1中的葡萄糖原料成本约为90元。以d-乳酸生产车间每年产1000吨d-乳酸计算,每年可节约生产成本100万元。
虽然实施例2.8中同样采用糖化液(淀粉糖)作为乳酸发酵的主要碳源,但是其淀粉糖制备过程中采用的是低浓度的淀粉乳作为原料。与实施例2.1相比,采用30重量%以上的高浓度淀粉质原料进行液化处理获取糖化液的方法,能够大幅缩短原料处理时间,从而提高了原料预处理效率。将其应用于乳酸发酵生产中时可以降低原料预处理操作成本,进而降低整体的生产成本,提高生产效率。
通过对比实施例2.1与参比实施例2.3-2.5的结果可以看出,只有在本发明提供的乳酸发酵条件下,配合本发明提供的方法,才能实现提高糖酸转化率和生产强度的同时,缩短发酵周期和降低生产成本的目的。
实施例3.1-3.7和参比实施例3.1-3.6
这些实施例用来说明本发明涉及的发酵制备乳酸的方法。其中:
在无特殊说明的情况下,采用的菌株为保藏编号为cgmccno.16833的乳酸片球菌,具体参见cn109536409a。-80℃超低温冰箱保存,甘油管每隔1-3个月活化复培一次。
在以下实施例中,所述乳酸片球菌新鲜种子液的配制方法为:将低温冻存的乳酸片球菌菌株接种于mrs液体培养基中,所述菌株的接入量以1-10体积%为宜。温度40±2℃,转速170±30rpm条件下过夜培养,得到新鲜种子液。
所述mrs液体培养基的配制方法为:葡萄糖200g/l,酵母提取物5g/l,固含40%的玉米浆10ml/l,乙酸钠2g/l,kh2po40.5g/l,mgso4·7h2o0.5g/l,mnso4·h2o0.25g/l,吐温1ml/l。
在以下实施例中,所述纤维素酶解液的制备方式包括:将纤维素原料经粉碎、硫酸(浓度为1.6重量%)酸解、蒸汽汽爆(条件160℃,40min)和纤维素酶(诺维信公司的cellicctec3纤维素复合酶)酶解后,经滤布挤压制成。具体制备方式如下:
1、将木质纤维素原料粉碎至粒度为50±10mm,投入螺旋喂料器,对其进行挤压脱水至干物含量40±5重量%,形成致密的料塞,在保证纤维素原料不断进入处理容器的同时还能抵御容器内的蒸汽外泄;
2、将所述料塞从螺旋喂料器处理后,向其中混入硫酸,以所述料塞的重量为基准,所述硫酸的加入量为2±1重量%,在酸混合罐中混合后得到酸性纤维素原料;
3、将酸性纤维素原料在蒸煮处理容器中与蒸汽进行接触,条件为压力0.6mpa,时间50min。然后按照连续排料的方式排出容器;
4、将所得产物进行水洗,调节ph至5±0.1,加热至50℃后,加入纤维素酶,以每克产物的干重计,所述纤维素酶的添加量为0.15g。在50℃下保温搅拌72h,获得纤维素酶解液。
在以下实施例中,所述玉米浆为购自中粮生化榆树公司的玉米浓浆产品。kh2po4、mgso4•7h2o、mnso4•h2o购自国药试剂公司。所述纤维素酶为购自诺维信公司的cellicctec3纤维素复合酶产品。未做特殊说明时,采用的发酵罐为赛多利斯公司的bio-stat型号的2-5l发酵罐。
在以下实施例中,糖酸转化率的计算方式为:发酵结束时刻的乳酸总重量/初始的葡萄糖和五碳糖总重量。五碳糖包括:木糖和阿拉伯糖。葡萄糖含量的测定方式为:生物传感分析仪(采用山东省科学院生物研究所sba-40d型号的生物传感分析仪)。高温灭菌的条件为120℃,20min。
在以下实施例中,所述五碳糖含量的测定方法为高效液相色谱法,其条件参照刘建伟.hplc法测定木糖母液的组成[j].安徽农业科学,v.37(05):1881-1882。
在以下实施例中,所述乳酸的测定方式为高效液相色谱法,其条件如下所述:
仪器设备:agilenttechnologies1260infinityii;
检出器:rid;
分离柱:aminexhpx-87hcolumn300×7.8mm;
柱温:55℃
流动相:0.005m硫酸;
流量:0.5ml/min;
进样量:20μl;
乳酸保留时间为14min左右。
实施例3.1
采用2l的发酵罐进行小试水平实验。
培养基制备:所述纤维素原料为秸秆。按照30体积%纤维素酶解液作为碳源,8体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至40℃待用。其中,五碳糖的含量为44g/l,葡萄糖的含量为100g/l。
预发酵:将1.2l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照5体积%接种量接入发酵罐(浓度为od600=0.45),温度控制42℃,发酵转速为150r/min,开启微量通气通入空气,通气量以氧气记为0.05vvm。以氢氧化钠溶液(浓度4重量%)为中和剂,在发酵过程中流加并控制ph为6。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以42℃,发酵转速为150r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,在发酵过程中流加并控制ph为6的条件进行乳酸发酵。
结果:总发酵时间为75h,其中,预发酵时间为15h,继续发酵时间为60h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为48%。总糖酸转化率达到78%,乳酸产量可达到95g/l。
实施例3.2
采用2l的发酵罐进行小试水平实验。
培养基制备:所述纤维素原料为秸秆。按照10体积%纤维素酶解液作为碳源,3体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至40℃待用。其中,五碳糖的含量为15g/l,葡萄糖的含量为36g/l。
预发酵:将1.2l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照1体积%接种量接入发酵罐(浓度为od600=0.09),温度控制37℃,发酵转速为150r/min,开启微量通气通入空气,通气量以氧气记为0.05vvm。以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为6.2。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以37℃,发酵转速为150r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为6.2的条件进行乳酸发酵。
结果:总发酵时间为48h,其中,预发酵时间为8h,继续发酵时间为40h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为53%。总糖酸转化率达到80%,乳酸产量可达到35g/l。
实施例3.3
采用2l的发酵罐进行小试水平实验。
培养基制备:所述纤维素原料为秸秆。按照20体积%纤维素酶解液作为碳源,5体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至40℃待用。其中,五碳糖的含量为30g/l,葡萄糖的含量为70g/l。
预发酵:将1.2l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照3体积%接种量接入发酵罐(浓度为od600=0.27),温度控制39℃,发酵转速为150r/min,开启微量通气通入空气,通气量以氧气记为0.05vvm。以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为6.5。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以39℃,发酵转速为100r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为6.5的条件进行乳酸发酵。
实验结果:总发酵时间为66h,其中,预发酵时间为15h,继续发酵时间为51h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为47%。总糖酸转化率达到79%,乳酸产量可达到66g/l。
实施例3.4
采用5l的发酵罐进行小试水平实验。
培养基制备:所述纤维素原料为燕麦壳。按照15体积%纤维素酶解液作为碳源,4体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至45℃待用。其中,五碳糖的含量为20g/l,葡萄糖的含量为55g/l。
预发酵:将3.0l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照7体积%接种量接入发酵罐(浓度为od600=0.63),温度控制45℃,发酵转速为50r/min,开启微量通气通入空气,通气量以氧气记为0.008vvm。以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为5.3。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以45℃,发酵转速为70r/min,以氨水溶液(浓度14重量%)为中和剂,过程中流加并控制ph为5.3的条件进行乳酸发酵。
结果:总发酵时间为55h,其中,预发酵时间为12h,继续发酵时间为43h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为51%。总糖酸转化率达到78%,乳酸产量可达到51g/l。
实施例3.5
采用2l的发酵罐进行小试水平实验。
培养基制备:所述纤维素原料为玉米芯。按照25体积%纤维素酶解液作为碳源,7体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至40℃待用。其中,五碳糖的含量为35g/l,葡萄糖的含量为85g/l。
预发酵:将1.2l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照5体积%接种量接入发酵罐(浓度为od600=0.45),温度控制40℃,发酵转速控制120r/min,开启微量通气通入空气,通气量以氧气记为0.01vvm。以氨水溶液(浓度12重量%)为中和剂,过程中流加并控制ph为6.5。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以40℃,发酵转速为120r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,过程中流加并控制ph为6.5的条件进行乳酸发酵。
结果:总发酵时间为75h,其中,预发酵时间为18h,继续发酵时间为57h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为52%。总糖酸转化率达到81%,乳酸产量可达到84g/l。
实施例3.6
采用100l的发酵罐进行中试水平实验,所述发酵罐为上海保兴生物工程有限公司的biotech-100js型号的100l发酵罐。
培养基制备:所述纤维素原料为秸秆。按照20体积%纤维素酶解液作为碳源,6体积%的玉米浆液作为氮源,并进一步添加无机盐kh2po41g/l、mgso4·7h2o0.5g/l、mnso4·h2o0.25g/l,余量为水,高温灭菌后冷却至45℃待用。其中,五碳糖的含量为30g/l,葡萄糖的含量为70g/l。
预发酵:将57l培养基加入发酵罐后,将乳酸片球菌新鲜种子液按照5体积%接种量接入发酵罐(浓度为od600=9),温度控制42℃,发酵转速控制80r/min,开启微量通气通入空气,通气量以氧气记为0.01vvm。以氨水溶液(浓度14%)为中和剂,过程中流加并控制ph为6.0。
继续发酵:当发酵体系中乳酸片球菌的含量达到od600=10时,停止通气,继续以42℃,发酵转速为50r/min,以氨水溶液(浓度14重量%)为中和剂,过程中流加并控制ph为6的条件进行乳酸发酵。
结果:总发酵时间为62h,其中,预发酵时间为14h,继续发酵时间为48h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为46%。总糖酸转化率达到78%,乳酸产量可达到65g/l。
实施例3.7
按照实施例3.1的方式,不同之处在于,采用柠檬酸钠溶液(浓度为11重量%)作为中和剂。
结果:总发酵时间为80h,其中,预发酵时间为20h,继续发酵时间为60h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为49%。总糖酸转化率达到80%,乳酸产量可达到97g/l。
实施例3.8
按照实施例3.1的方式,不同之处在于,采用如下方式进行继续发酵:
当发酵体系中乳酸片球菌的含量达到od600=8时,停止通气,继续以42℃,发酵转速为150r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,在发酵过程中流加并控制ph为6的条件进行乳酸发酵。
结果:总发酵时间为70h,其中,预发酵时间为12h,继续发酵时间为58h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为48%。总糖酸转化率达到72%,乳酸产量可达到90g/l。
实施例3.9
按照实施例3.1的方式,不同之处在于,采用如下方式进行继续发酵:
当发酵体系中乳酸片球菌的含量达到od600=13时,停止通气,继续以42℃,发酵转速为150r/min,以氢氧化钠溶液(浓度4重量%)为中和剂,在发酵过程中流加并控制ph为6的条件进行乳酸发酵。
结果:总发酵时间为72h,其中,预发酵时间为16h,继续发酵时间为56h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为50%。总糖酸转化率达到77%,乳酸产量可达到93g/l。
参比实施例3.1
采用实施例3.1的方式,不同之处在于,采用全程厌氧发酵的方式,即,预发酵和继续发酵过程中均不通入空气。
结果:总发酵时间为90h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为42%。总糖酸转化率达到73%,乳酸产量可达到86g/l。
参比实施例3.2
采用实施例3.1的方式,不同之处在于,预发酵过程中通气量以氧气记为0.1vvm。
结果:总发酵时间为72h,其中,预发酵时间为12h,继续发酵时间为60h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为52%。总糖酸转化率达到66%,乳酸产量可达到81g/l。
参比实施例3.3
采用实施例3.1的方式,不同之处在于,接种的乳酸片球菌的种子液接种量为1体积‰。
结果:总发酵时间为90h,其中,预发酵时间为20h,继续发酵时间为70h。发酵结束后,无葡萄糖残留,五碳糖消耗量为33%。总糖酸转化率达到66%,乳酸产量可达到76g/l。
参比实施例3.4
采用实施例3.1的方式,不同之处在于,当培养基中的五碳糖消耗达到60%再停止发酵。
结果:总发酵时间为120h,其中,预发酵时间为15h,继续发酵时间为105h。发酵结束后,无葡萄糖残留,五碳糖消耗量为60%。总糖酸转化率达到69%,乳酸产量可达到87g/l。
参比实施例3.5
采用实施例3.1的方式,不同之处在于,预发酵后,发酵体系内乳酸片球菌生物量od600≥20。
结果:总发酵时间为68h,其中,预发酵时间为18h,继续发酵时间为50h。发酵结束后,无葡萄糖残留,五碳糖消耗量为49%。总糖酸转化率达到66%,乳酸产量可达到80g/l。
参比实施例3.6
采用实施例3.1的方式,不同之处在于,在预发酵和继续发酵过程中均通入空气,即采用全程通气的方式通入空气。
结果:总发酵时间为66h,其中,预发酵时间为14h,继续发酵时间为52h。发酵结束后,无明显葡萄糖残留,五碳糖消耗量为56%。总糖酸转化率达到54%,乳酸产量可达到67g/l。
通过实施例3.1与参比实施例3.1的结果对比可以看出,采用本发明提供的优选实施方式,即在预发酵阶段通入空气的方式,在乳酸生产应用中的总发酵时间更少,节约了时间成本。同时,发酵培养基中的五碳糖消耗量、糖酸转化率和乳酸产量均有所增加,提高了生产效率。
通过实施例3.1与参比实施例3.2的结果对比可以看出,虽然氧气通入量增加可以略微减少总发酵时间和培养基中五碳糖的消耗量,但是其糖酸转化率和乳酸产量明显下降。从这个结果可以看出,增加的五碳糖消耗并非用于乳酸发酵,而是更多供应菌体生长,对于乳酸发酵生产的生产效率无益。
通过实施例3.1与参比实施例3.3的结果对比可以看出,当乳酸片球菌的接种量低于本发明优选的接种量范围时,在乳酸发酵生产过程中的总发酵时间增加,培养基中五碳糖的消耗量、糖酸转化率和乳酸产量均有所下降。总体的生产效率降低,不利于工业化规模生产应用。
通过实施例3.1与参比实施例3.4的结果对比可以看出,当培养基中五碳糖的消耗量超过本发明优选的消耗量范围时,随着总发酵时间的增加,糖酸转化率和乳酸产量并没有相应增加。并且造成总体的生产效率较低,不利于工业化规模生产应用。
通过实施例3.1与参比实施例3.5的结果对比可以看出,当预发酵阶段结束时发酵体系内乳酸片球菌过多时,虽然总发酵时间有所降低,但是其中的预发酵时间相应增加,且总糖酸转化率和乳酸产量均有所下降,总体的生产效率降低。结合五碳糖的消耗量有所增加的结果看,可能是发酵体系中的营养成分更多供应了菌体生长而非乳酸生产。
通过实施例3.1与参比实施例3.6的结果对比可以看出,虽然发酵时间有所减少,但是总糖酸转化率和乳酸产量均有所下降。根据五碳糖消耗量的对比看,全程通气的条件下,发酵体系中的营养成分更多供应了菌体生长而非乳酸生产,造成生产效率下降,不利于工业化规模生产应用。
实施例4.1-4.5和参比实施例4.1-4.2
这些实施例用来说明本发明涉及的分离乳酸的方法。其中:
乳酸发酵菌种为鼠李糖乳杆菌,cgmccno.16834(已公开于cn109628339a)。
旋蒸仪购买于上海亚荣生化仪器厂,仪器型号为re-52aa。
刮膜蒸发器购买于uic公司,仪器型号为gmbh。
乳酸的含量通过高效液相色谱法测试得到。
实施例和参比实施例制备精制乳酸的参数均列于表1。
实施例4.1
(1)将鼠李糖乳杆菌的乳酸发酵液(乳酸含量16.6重量%)依次进行固液分离和脱色,其中,脱色剂为活性炭;
(2)将步骤(1)得到的产物与阴离子交换树脂西安蓝晓lsd296按体积比1:2进行离子交换,得到乳酸溶液;
(3)将步骤(2)得到的乳酸浓液在旋蒸仪中进行减压浓缩,得到乳酸含量为85重量%、水含量为15重量%的乳酸浓液,其中,所述减压浓缩的条件包括:温度为40℃,真空度为0.7mbar;
(4)将步骤(3)得到的乳酸浓液在刮膜蒸发器中进行分子蒸馏,得到乳酸含量为99重量%的精制乳酸s1,其中,所述刮膜蒸发器的条件包括:刮板转速为70r/min,蒸汽夹套内部温度为110℃,进料流速为10ml/min,真空度为0.4mbar。
实施例4.2
(1)将鼠李糖乳杆菌的乳酸发酵液(乳酸含量14.5重量%)依次进行固液分离和脱色,其中,脱色剂为活性炭;
(2)将步骤(1)得到的产物与阴离子交换树脂西安蓝晓lsa-700b按体积比1:5进行离子交换,得到乳酸溶液;
(3)将步骤(2)得到的乳酸浓液在旋蒸仪中进行减压浓缩,得到乳酸含量为80重量%、水含量为20重量%的乳酸浓液,其中,所述减压浓缩的条件包括:温度为60℃,真空度为0.8mbar;
(4)将步骤(3)得到的乳酸浓液在刮膜蒸发器中进行分子蒸馏,得到乳酸含量为98重量%的精制乳酸s2,其中,所述刮膜蒸发器的条件包括:刮板转速为90r/min,蒸汽夹套内部温度为120℃,进料流速为12ml/min,真空度为0.1mbar。
实施例4.3
(1)将鼠李糖乳杆菌的乳酸发酵液(乳酸含量14.4重量%)依次进行固液分离和脱色,其中,脱色剂为活性炭;
(2)将步骤(1)得到的产物与阴离子交换树脂西安蓝晓d303按体积比1:7进行离子交换,得到乳酸溶液;
(3)将步骤(2)得到的乳酸浓液在旋蒸仪中进行减压浓缩,得到乳酸含量为80重量%、水含量为20重量%的乳酸浓液,其中,所述减压浓缩的条件包括:温度为30℃,真空度为0.8mbar;
(4)将步骤(3)得到的乳酸浓液在刮膜蒸发器中进行分子蒸馏,得到乳酸含量为99重量%的精制乳酸s3,其中,所述刮膜蒸发器的条件包括:刮板转速为100r/min,蒸汽夹套内部温度为110℃,进料流速为10ml/min,真空度为0.08mbar。
实施例4.4
按照实施例4.1的方法,不同的是,将鼠李糖乳杆菌的乳酸发酵液(乳酸含量16.6重量%)替换为乳酸含量为13.3重量%的含乳酸溶液,得到精制乳酸s4。
实施例4.5
按照实施例4.1的方法,不同的是,省略步骤(1),即:直接将鼠李糖乳杆菌的乳酸发酵液(乳酸含量16.6重量%)进行离子交换,得到乳酸精品s5。
参比实施例4.1
将鼠李糖乳杆菌的乳酸发酵液(乳酸含量16.6重量%)按照cn103724183a公开的方法进行分离,得到精制乳酸d1。
参比实施例4.2
按照实施例4.1的方法,不同的是,省略步骤(3),将步骤(2)得到的乳酸浓液直接进行分子蒸馏,得到精制乳酸d2。
表1
通过表1数据可知,采用本发明提供的方法,将含乳酸溶液进行离子交换、减压浓缩和分子蒸馏处理,有利于提高精制乳酸中乳酸的含量,提高了乳酸的纯度。
实施例5.1-5.6和参比实施例5.1-5.4
这些实施例用来说明本发明涉及的分离乳酸盐的方法和系统。其中:
板框过滤机购自海宁市云飞过滤设备有限公司,型号为yf-100-1。
移动式空气压缩机购自上海国厦压缩机有限公司,型号为w1.5/20。
液体增压泵购自安徽腾龙泵阀制造有限公司,型号为cqb32-25-125f。
乳酸盐检测方法,采用高效液相色谱法检测法;方法如下:色谱仪:agilenttechnologies1260infinityii;检出器:rid;分离柱:aminexhpx-87hcolumn300×7.8mm;流动相:0.005m硫酸;流量:0.5ml/min;进样量:20μl;乳酸盐保留时间为10-20min。
乳酸发酵菌种为鼠李糖乳杆菌,cgmccno.16834(cn109628339a)。
乳酸盐发酵液的制备方法:将乳酸菌接种至含有碳氮源等成分的发酵培养基中进行乳酸发酵,发酵过程中控制温度37-45℃,ph=5.5-6.3,低速搅拌,直至底料中碳源消耗殆尽,发酵结束,获得新鲜的发酵液;其中,所述乳酸盐发酵液中,以乳酸盐铵计的乳酸盐的含量为150g/ml。
下游处理是指浓缩,即:先用离子交换脱除阳离子和阴离子,再分子蒸馏除去水分。
进行下游处理的液体体积、乳酸盐含量以及乳酸盐收率均列于表2。
实施例5.1
(1)将1000l乳酸盐发酵液和20g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为20℃、转速50rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.2mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为800目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.15mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡60min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.15mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s1。
实施例5.2
(1)将1000l乳酸盐发酵液和10g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为30℃、转速20rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.3mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为500目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.1mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡30min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.1mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s2。
实施例5.3
(1)将1000l乳酸盐发酵液和30g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为30℃、转速20rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.4mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为1000目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.25mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡90min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.25mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s3。
实施例5.4
(1)将1000l乳酸盐发酵液和60g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为0℃、转速100rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.35mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为600目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.1mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡60min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.05mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s5。
实施例5.5
(1)将1000l乳酸盐发酵液和100g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为40℃、转速20rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.05mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为800目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.05mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡120min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.05mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s5。
实施例5.6
(1)将1000l乳酸盐发酵液和5g珍珠岩注入分离单元中带有搅拌装置的混合罐中,在温度为10℃、转速50rpm的条件下进行搅拌混合,直至珍珠岩完全悬浮在发酵液中,得到混合物料;
(2)将所述混合物料通过增压泵注入板框过滤机中,在压力为0.45mpa、搅拌的情况下进行过滤,直至混合物料全部滤完,关掉增压泵,得到第一料液和滤饼,其中,所述板框过滤中滤布的目数为500目;
(3)过滤结束后,在板框过滤机的进液口通入压力为0.3mpa的压缩空气,吹顶出残留在滤饼中的乳酸盐透过液,直至吹顶出的液体非常少,且压缩空气中仅有少量液沫夹带,停止第一顶吹;
(4)第一顶吹结束后,在板框过滤机的进液口通入水,直至出口处流水呈连续状,关闭进口阀和出口阀,浸泡30min,水浸泡结束后,得到菌体细胞中乳酸盐残留液;
(5)水洗结束后,再在板框过滤机的进液口通入压力为0.3mpa的压缩空气进行第二顶吹,置换出滤饼中的水洗液;
(6)将所述第一料液、乳酸盐透过液、乳酸盐残留液和水洗液的混合液进行下游处理,得到乳酸盐s6。
参比实施例5.1
按照cn104557515a的方法,用离心分离代替实施例5.1中的气顶水洗,将离心清液进行下游处理,得到乳酸盐d1。
参比实施例5.2
按照cn104557515a的方法,用离心分离代替实施例5.1中的气顶水洗,并将离心后的固相用等体积的水洗两次,将水洗液和离心清液混合,然后进行下游处理,得到乳酸盐d2。
参比实施例5.3
按照实施例5.1的方法,删除步骤(3)-(5),直接将步骤(2)得到的滤液进行下游处理,得到乳酸盐d3。
参比实施例5.4
按照实施例5.1的方法,删除步骤(3)和(5),即:直接将步骤(2)得到的滤液和步骤(4)得到的滤饼洗液进行下游处理,得到乳酸盐d4。
表2
通过表2的结果可以看出,相比参比实施例5.1-5.2,采用本发明提供的方法具有较高的乳酸盐含量和乳酸盐收率,参比实施例5.3不进行气顶水洗,虽然乳酸盐含量与发酵液相近,但收率只有84%;参比实施例5.4不进行气顶,仅采用水洗,虽然收率接近89%,但溶液过于稀释,乳酸盐的含量较低。因此,相比参比实施例5.1-5.4,采用本发明提供的分离乳酸盐的方法在产业化过程分离效果较高,成本较低。
实施例6.1-6.12和参比实施例6.1-6.5
这些实施例在于说明采用本发明涉及的装置和方法制备的聚乳酸。其中:
(1)pla切片色泽是最直观反映pla切片质量的指标,并且在一定程度上影响下游制品的色彩。色泽是光泽(消光程度)和颜色(黄色程度)的总称,是反射光线在空间分布、波谱分布及方向性上的反映。色泽的测量是根据色度学与光度学原理及国际照明委员会(cie)计量标准,通常用亨特(l、a、b)法色差计进行测量,其结果用l值、a值、b值表示。l值表示白度,其值越大,则亮度高、白度大;a值大表示红色指数大;b值大表示黄色指数大。色泽参数通过cm-5分光测色计测得,其中,cm-5分光测色计购自日本柯尼卡美能达公司;
(2)使用气相色谱仪gc580检测聚乳酸切片单体含量,其中,气相色谱仪gc580购自美国珀金埃尔默公司;
(3)力学性能测试
在发明中,拉伸强度和断裂伸长率采用智能电子拉力试验机准进行测试,仪器购自济南兰光机电电子有限公司,型号为xlw。
(4)重均分子量
在本发明中,重均分子量采用凝胶渗透色谱仪测试。
(5)原料来源
受阻酚类抗氧剂和亚磷酸酯类抗氧剂均可通过商购获得,购自上海迈瑞尔化学技术有限公司。
实施例6.1
(1)丙交酯熔融工序:在连续地氮气的保护下,将丙交酯加入到丙交酯熔融罐i中,在95℃下熔融1h,得到熔融态的丙交酯;
(2)第一次聚合反应工序:将熔融态的丙交酯通入第一次聚合反应器ii,同时添加催化剂辛酸亚锡、开环聚合引发剂聚乙二醇、高效复合稳定剂cs-12(复合稳定剂cs-12是由at-76与at-626按质量比2:1复配),在温度为155℃,压力为50kpa,反应时间为3h的条件下进行第一次聚合反应,转化率达到55%。
(3)第二次聚合反应工序:将所述第一次聚合反应后的第一熔体通入第二次聚合反应器iii,在温度为195℃,压力为6mpa,反应时间为1h的条件下进行第二次聚合反应,转化率达到95%;
(4)脱单体工序:将所述第二次聚合反应器内的第二熔体通入脱单反应器iv,并加入终止剂亚磷酸,进行脱单体反应,其中,激振器振幅a=0.15mm,振频f=25hz,温度为210℃,压力为1kpa,反应时间为0.5h的条件下进行,最终得到的聚乳酸残留丙交酯单体含量为4.1‰;
(5)造粒工序:将聚乳酸熔体连续地经熔体管道、送料泵通入双螺杆挤出机v,经水冷切粒、脱水、筛选、结晶、除湿干燥,最终得到聚乳酸树脂,造粒机温度为210℃。
实施例6.2
(1)丙交酯熔融工序:连续地氮气的保护下,将丙交酯加入到丙交酯熔融罐i中,在90℃下熔融1h,得到熔融态的丙交酯;
(2)第一次聚合反应工序:将熔融态的丙交酯通入第一次聚合反应器ii,同时添加催化剂辛酸亚锡、开环聚合引发剂聚乙二醇、高效复合稳定剂cs-9(复合稳定剂cs-9是由at-10与at-168按质量比2:1复配),在温度为150℃,压力为50kpa,反应时间为3h的条件下进行第一次聚合反应,转化率达到55%;
(3)第二次聚合反应工序:将所述第一次聚合反应后的第一熔体通入第二次聚合反应器iii,在温度为190℃,压力为6mpa,反应时间为1h的条件下进行第二次聚合反应,转化率达到95%;
(4)脱单体工序:将所述第二次聚合反应器内的第二熔体通入脱单反应器iv,并加入终止剂亚磷酸,进行脱单体反应,其中,激振器振幅a=0.25mm,振频f=30hz,温度为215℃,压力为1kpa,反应时间为0.5h的条件下进行,最终得到的聚乳酸残留丙交酯单体含量为0.8‰;
(5)造粒工序:将聚乳酸熔体连续地经熔体管道、送料泵通入双螺杆挤出机v,经水冷切粒、脱水、筛选、结晶、除湿干燥,最终得到聚乳酸树脂,造粒机温度为205℃。
实施例6.3-6.12
按照与实施例6.1相同的装置和方法制备聚乳酸,所不同之处在于:将复合稳定剂cs1-12进行替换,以及将步骤(d)中的条件进行修改,制备的条件如表3所示,以及测试结果如表4所示。
参比实施例6.1
采用常规pla切片,重均分子量为15万,熔融指数6g/10min,色泽中l值75.15、a值-1.13、b值19.08,单体含量5.2‰。
参比实施例6.2
按照与实施例6.1相同的装置和方法制备聚乳酸,所不同之处在于:将复合稳定剂cs1-12替换为d-2,该d-2是由at-76与at-626按质量比1:3复配;测试结果如表4所示。
参比实施例6.3
按照与实施例6.1相同的装置和方法制备聚乳酸,所不同之处在于:在步骤(4)中,激振器振幅a=0mm,振频f=0hz;测试结果如表4所示。
参比实施例6.4
按照与实施例6.1相同的装置和方法制备聚乳酸,所不同之处在于:没有进行步骤(3);测试结果如表4所示。
参比实施例6.5
按照与实施例6.1相同的装置和方法制备聚乳酸,所不同之处在于:丙交酯、催化剂、高效复合稳定剂、引发剂、以及终止剂的用量质量比分别为1:0.006:0.003:0.007:0.006;测试结果如表4所示。
表3
测试例1
取实施例6.1-6.12和参比实施例6.1-6.5样品,使用cm-5分光测色计检测色值,测试结果见表4所示,表4为pla切片色值和残留丙交酯单体含量。
测试例2
取实施例6.1-6.12和参比实施例6.1-6.5样品,使用气相色谱仪gc580检测聚乳酸切片单体含量,测试结果见表4所示。
表4
由表4可以看出:
(1)从实施例6.1-6.12中pla聚合过程中使用不同复合稳定剂制备的pla切片色值与参比实施例6.1采用现有常规pla切片以及参比实施例6.2-6.5制备的pla切片色值数据对比上看:
其中,参比实施例6.1现有常规pla切片的l值为75.15,b值为19.08;参比实施例6.2-6.5由于在pla聚合过程中没有采用本发明的技术方案中所限定的条件,则结果pla切片b值较高,黄度大。
而实施例6.1-6.12,加入复合稳定剂后,pla切片的b值明显降低,说明添加复合稳定剂后,聚合反应具有较高的热稳定性及抗热氧化性,得以获得色值指标较好的pla切片。另外,从表4中还可以看到,实施例6.2中添加复合稳定剂cs-9的pla切片b值仅为4.19,效果最好。
(2)从实施例6.1-6.12制备的聚乳酸中单体含量与参比实施例6.1采用现有常规pla切片以及参比实施例6.2-6.5制备的聚乳酸中单体含量数据对比上看:
其中,参比实施例6.1中,pla切片的残留丙交酯单体含量为5.2‰;
其中,参比实施例6.2中,pla切片的残留丙交酯单体含量为4.2‰;
其中,参比实施例6.3脱单体工序中,电磁激振器振幅a=0mm、振频f=0hz,pla切片的残留丙交酯单体含量为4.1‰;
其中,参比实施例6.4-6.5中,pla切片的残留丙交酯单体含量为4.2-4.3‰;
而实施例6.1-6.12脱单体工序中,电磁激振器振幅a=0.15-0.3mm、振频f=20-45hz,pla切片的残留丙交酯单体含量最低能达到0.8‰。
因此,实施例6.1-6.12的结果说明振动力场的引入能够提高聚乳酸熔体的剪切速率,使熔体界面拉伸、压缩,产生相应的振荡,提高丙交酯单体挥发量,满足快速脱除单体的要求,将残留聚乳酸熔体的丙交酯单体含量控制在最低程度。
实施例7.1-7.7和参比实施例7.1-7.6
这些实施例用来说明本发明涉及的聚乳酸熔体在线制备改性聚乳酸材料的装置和方法。其中:
(1)力学性能测试
在发明中,拉伸强度和断裂伸长率采用智能电子拉力试验机准进行测试,仪器购自济南兰光机电电子有限公司,型号为xlw。
(2)重均分子量
在本发明中,重均分子量采用凝胶渗透色谱仪测试。
(2)原料来源
聚己二酸对苯二甲酸丁二醇酯购自新疆蓝山屯河化工股份有限公司;
聚丙撑碳酸酯购自内蒙古蒙西高新材料股份有限公司;
聚丁二酸丁二醇酯购自巴斯夫股份公司;
聚丁二醇-丁二酸/己二酸共聚酯购自金发科技股份有限公司;
滑石粉购自上海诚致化工有限公司;
碳酸钙购自浙江天石纳米科技有限公司;
柠檬酸三丁酯购自上海迈瑞尔化学技术有限公司;
环氧大豆油购自东莞市恒泰化工有限公司;
己二酸二乙二醇单丁醚酯购自长春应用化学研究所;
扩链剂购自巴斯夫股份公司;
交联剂购自东莞涣宗贸易有限公司;
抗氧剂购自天津利安隆新材料有限公司;
润滑剂购自四川天宇油脂化学有限公司。
实施例7.1
本实施例在于说明采用本发明的装置和方法制备的改性聚乳酸材料。
如图8所示:
(a)将聚合后的59.1重量份聚乳酸熔体经聚乳酸熔体管道连续地输出进入聚乳酸熔体进料计量泵10c(熔体进料计量泵)连续的输出进入双螺杆挤出机的四区;
(b)将35重量份第一固体改性剂聚己二酸对苯二甲酸丁二醇酯和5重量份第二固体改性剂滑石粉分别加入第一固体改性剂料斗8c和第二固体改性剂料斗9c中;
(c)将0.3重量份液体改性剂柠檬酸三丁酯通过液体进料计量泵12c以侧线喂料的方式进入双螺杆挤出机二区;
(d)将0.1重量份扩链剂、0.1重量份交联剂、0.2重量份抗氧剂、0.2重量份润滑剂混合均匀后加入辅助料斗6c中,并将制备得到100重量份的改性聚乳酸聚合熔体在双螺杆挤出机五区至十一区混合均匀挤出造粒。
结果制备得到改性聚乳酸材料。
实施例7.2
本实施例在于说明采用本发明的装置和方法制备的改性聚乳酸材料。
(a)将聚合后的53重量份聚乳酸熔体经聚乳酸熔体管道连续地输出进入聚乳酸熔体进料计量泵10c(熔体进料计量泵)连续的输出进入双螺杆挤出机的四区;
(b)将39重量份第一固体改性剂聚己二酸对苯二甲酸丁二醇酯和7重量份第二固体改性剂滑石粉分别加入第一固体改性剂料斗8c和第二固体改性剂料斗9c中;
(c)将0.2重量份液体改性剂环氧大豆油通过液体进料计量泵12c以侧线喂料的方式进入双螺杆挤出机二区;
(d)将0.2重量份扩链剂、0.1重量份交联剂、0.3重量份抗氧剂、0.2重量份润滑剂混合均匀后加入辅助料斗6c中,并将制备得到100重量份的改性聚乳酸聚合熔体在双螺杆挤出机五区至十一区混合均匀挤出造粒。
结果制备得到改性聚乳酸材料。
实施例7.3-7.7
按照与实施例7.1相同的装置和方法制备的改性聚乳酸材料,所不同的是,相应修改实施例7.1中的第一固体改性剂、第二固体改性剂、液体改性剂、扩链剂、交联剂、抗氧剂和润滑剂,具体如表5所示。
表5
参比实施例7.1
按照与实施例7.2相同的方法制备改性聚乳酸材料,所不同的是,采用中粮吹膜级聚乳酸切片,按照实施例7.2的配方,将53重量份聚乳酸切片与39重量份聚己二酸对苯二甲酸丁二醇酯、7重量份滑石粉、0.2重量份环氧大豆油、0.2重量份扩链剂、0.1重量份交联剂、0.3重量份抗氧剂、0.2重量份润滑剂机械混合均匀后通过双螺杆挤出机挤出造粒。
结果制备得到改性聚乳酸材料。
参比实施例7.2-7.6
按照与实施例7.1相同的装置和方法制备的改性聚乳酸材料,所不同的是,聚乳酸熔体的含量以及第一固体改性剂和第二固体改性剂的含量不一样,具体如表6所示。
表6
测试例3
将实施例7.1-7.7和参比实施例7.1-7.6制备的改性聚乳酸材料拉伸强度和断裂伸长率,结果如表7所示。
表7
通过表7的结果可以看出,采用本发明的实施例7.1-7.7制备的聚乳酸改性材料具有更好的性能,其中,采用本发明中实施例7.2制备的聚乳酸改性材料具有更好的性能,其分子量为184059g/mol,拉伸强度为56.2mpa,断裂伸长率为125%。
实施例8.1-8.2和参比实施例8.1
这些实施例用来说明本发明涉及的聚乳酸脱挥蒸发器。
实施例8.1
将丙交酯通过熔融聚合法得到的转化率为94.6%的聚乳酸/丙交酯物料以10kg/h的流量通过包括如图9所示的脱挥装置进行残留单体脱除。
该实施例中,搅拌带6的外沿和筒体3内壁之间的间隙为25mm,搅拌带6的螺距为筒体3内径的0.5倍,搅拌带6的宽度为筒体3内径的0.075倍,搅拌轴5的转速为15转/分,通入夹套4的水蒸汽温度为200℃,蒸发器内绝对压力为8kpa,
测得薄膜蒸发器出料口处的聚乳酸切片中单体的含量为0.16wt%。
实施例8.2
将丙交酯通过熔融聚合法得到的转化率为95.8%的聚乳酸/丙交酯物料以10kg/h的流量通过如图10所示的脱挥装置进行残留单体脱除。
该实施例中,搅拌带6的外沿和筒体3内壁之间的间隙为5mm,搅拌带6的螺距为筒体3内径的0.75倍,搅拌带6的宽度为筒体3内径的0.06倍,搅拌轴5的转速为45转/分,通入夹套4的导热油温度为220℃,蒸发器内绝对压力为2kpa。
测得蒸发器出料口处的聚乳酸切片中单体的含量为0.09wt%。
参比实施例8.1
将丙交酯通过熔融聚合法得到的转化率为95.2%的聚乳酸/丙交酯物料以10kg/h的流量通过中国实用新型专利cn204932896u提供的脱挥装置进行残留单体脱除。
参比实施例中筒体内壁和搅拌轴外侧的刮刀之间的间距为5mm。在搅拌轴的转速为30转/分,通入夹套的导热油温度为200℃,蒸发器内绝对压力为5kpa的操作条件下,测得薄膜蒸发器出料口处的聚乳酸切片中单体的含量为0.32wt%;将搅拌轴的转速提高到45转/分,通入夹套4的导热油温度提高到220℃,蒸发器内绝对压力减小到2kpa时,因聚乳酸物料在蒸发器的刮板上粘附挂壁严重,导致系统无法正常操作。
由实施例8.1-8.2与参比实施例8.1的对比可知,采用本发明提供的脱挥装置进行聚乳酸/丙交酯脱挥,可以将聚乳酸中单体的含量降至0.2wt%甚至0.1wt%以下,技术效果明显优于现有技术。
实施例9.1-9.5
这些实施例用于说明本发明涉及的丙交酯的制备方法
对乳酸原料分别进行预热、一级浓缩、二级浓缩、预缩聚、缩聚和解聚(以辛酸亚锡为催化剂),以进行丙交酯的连续化生产,其中,在各阶段中的条件如表8所示,最终得到的丙交酯产率和光纯度如表9所示。
表8
表9
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!