应用MLPA检测细胞遗传学异常的方法及试剂盒与流程
应用mlpa检测细胞遗传学异常的方法及试剂盒
技术领域
1.本发明属于细胞遗传学异常检测技术领域,尤其涉及应用mlpa检测细胞遗传学异常的方法及试剂盒。
背景技术:
2.多发性骨髓瘤(mm)是一种具有遗传学异质性的浆细胞恶性肿瘤,发病率占恶性血液病的10%~15%,好发于中老年人。主要表现为骨痛、贫血、持续感染、高钙血症、肾功能异常等。mm生存期从数月到15年以上不等,虽然近年来出现了一些新药使患者预后显著改善,但仍然不可治愈。因此建立准确的预后判断体系对制定精准治疗方案有重要的临床意义。
3.近年研究发现几乎所有的mm患者均存在细胞遗传学异常,遗传学异常是影响mm预后最重要的因素。对于细胞遗传学异常的检测方法主要包括传统染色体检测法、荧光原位杂交法、比较基因组杂交法和全基因组测序法。
4.传统染色体检测法基于显带技术,是将处理后的染色体染色,每个染色体都有特定带纹,带纹的变化表示该染色体的改变。其可以检测染色体数目及结构异常,对判定疾病危险度有重要临床意义。传统染色体检测必须在细胞中期分裂条件下进行,只能检出1/3患者的细胞遗传学异常,且由于结果判定依赖于检验者的经验,主观性大,在应用中受到了很大限制。
5.荧光原位杂交(fish)的基本原理是将寡聚核苷酸探针与变性后核酸按照碱基互补配对原则进行杂交,经处理后形成靶dna与核酸探针的杂交体,在荧光显微镜下显影,即可对待测dna进行定性、定量或相对定位分析。该技术不需要中期分裂象,检出率高,结果准确,检验快速,操作安全,成为传统染色体检测法的一个有益补充。但其局限性是不能检测完整的染色体变异,仅能辨识特异性探针结合区域。
6.比较基因组杂交(cgh)是在1992年后发展起来的分子细胞遗传学技术,其基本原理是用不同的荧光染料通过缺口平移法分别标记肿瘤组织和正常细胞或组织的dna,制成探针,并与健康人的间期染色体进行共杂交,以在染色体上显示肿瘤与健康对照的荧光强度的不同来反映整个肿瘤基因组dna表达情况的变化,再借助于图像分析技术可对染色体拷贝数量的变化进行定量研究。该法所需dna样本量较少,适用于外周血、培养细胞、新鲜组织样本、存档组织研究及dna量少而经聚合酶链反应(pcr)扩增样本的研究。但cgh技术所能检测到的最小的dna扩增或丢失是在3~5mb,故对于低水平的dna扩增和小片段的丢失会漏检;且价格较昂贵,有一定技术难度,目前主要应用于临床试验或科研,尚未普遍应用于临床。
7.全基因组测序即对一种生物的基因组的全部基因进行测序,测定其dna的碱基序列。该方法是在1986年首次被renato dulbecco提出,他认为如果能够知道所有人类基因序列,对癌症研究将有很大帮助。但该方法耗时、耗力、耗财,目前尚未用于临床。
8.随着检测技术的不断进步,遗传学异常的检出率逐渐提高。虽然目前fish是细胞
遗传学异常检测的标准方法,但仍有缺陷,并不能获得完整的染色体异常信息,检出率未达到遗传学异常的发生率,并且研究者水平参差不一,使得各个遗传学异常的预后意义仍存在差异。
技术实现要素:
9.有鉴于此,本发明的目的在于提供应用mlpa检测细胞遗传学异常的方法及试剂盒;本发明所述方法可以同时检测多达100个基因的外显子情况,包括小拷贝数的缺失或重复。并且所述试剂盒检测快速、简单、价格便宜、特异性高,对细胞遗传学检测有临床推广潜力。
10.为了实现上述发明目的,本发明提供了以下技术方案:
11.本发明提供了一种应用mlpa检测细胞遗传学异常的方法,包括以下步骤:
12.1)分别分离细胞检测样本和细胞参照样本中的单个核细胞,制备为细胞悬液;
13.2)将所述细胞悬液与cd138磁珠混合分选获得cd138阳性浆细胞;
14.3)提取所述cd138阳性浆细胞的基因组dna;
15.4)将所述基因组dna变性后,与mlpabuffer和mplaprobe mix混合进行mlpa反应获得mlpa产物;
16.5)将所述mlpa产物与连接酶混合液混合进行连接获得连接产物;
17.6)将所述连接产物和pcr聚合酶混合液混合进行pcr扩增获得pcr扩增产物;
18.7)对所述pcr扩增产物进行毛细管电泳,比较细胞检测样本和细胞参照样本的毛细管电泳结果分析细胞遗传学异常;当所述细胞检测样本的毛细管电泳结果在所述细胞参照样本的毛细管电泳结果范围内,认为细胞遗传学正常,否则认为细胞遗传学异常。
19.优选的,步骤3)中所述的基因组dna的a260/a280为1.6~1.9;所述基因组dna的浓度为20~30ng/μl。
20.优选的,步骤4)中所述变性的程序为98℃,5min变性;25℃,10min;所述变性后的基因组dna、mlpabuffer和mplaprobe mix的体积比为5:1.5:1.5。
21.优选的,所述mlpa反应的程序为95℃,1min;60℃,16~20h。
22.优选的,所述mplaprobe mix包括核苷酸序列如seq id no.1~seq id no.53所示的探针。
23.优选的,所述mlpa产物与连接酶混合液的体积比为1:4;所述连接的程序为54℃,15min;98℃,5min;20℃保持。
24.优选的,步骤6)所述的连接产物和pcr聚合酶混合液的体积比为4:1;所述pcr扩增的程序如下:95℃、30s;60℃、30s,72℃、60s,35个循环;72℃、20min;15℃停止。
25.优选的,所述细胞检测样本和细胞参照样本的数量比为(7~15):1。
26.本发明提供了应用mlpa检测细胞遗传学异常的试剂盒,包括所述的mplaprobe mix。
27.优选的,包括所述的pcr聚合酶混合液;所述pcr聚合酶混合液包括pcr扩增引物。
28.本发明的有益效果:本发明提供的应用mlpa检测细胞遗传学异常的方法,将mlpa应用于检测细胞遗传学异常,具有高通量、样本需求量少、耗时短、操作简单、重复性和灵敏度高的优势;能够更加快速、精确地检测细胞遗传学异常。
附图说明
29.图1为伴1q21扩增的mm患者mlpa结果;
30.图2为同时伴1q21扩增、13q缺失、17p缺失、16q缺失复杂细胞遗传学异常的mm患者mlpa结果;
31.图3为mlpa检测mm细胞遗传学异常数;
32.图4为mlpa检测不同遗传学异常的预后意义;
33.图5为mlpa检测细胞遗传学异常的流程。
具体实施方式
34.本发明提供了一种应用mlpa检测细胞遗传学异常的方法,包括以下步骤:1)分别分离细胞检测样本和细胞参照样本中的单个核细胞,制备为细胞悬液;2)将所述细胞悬液与cd138磁珠混合分选获得cd138阳性浆细胞;3)提取所述cd138阳性浆细胞的基因组dna;4)将所述基因组dna变性后,与mlpabuffer和mplaprobe mix混合进行mlpa反应获得mlpa产物;5)将所述mlpa产物与连接酶混合液混合进行连接获得连接产物;6)将所述连接产物和pcr聚合酶混合液混合进行pcr扩增获得pcr扩增产物;7)对所述pcr扩增产物进行毛细管电泳,比较细胞检测样本和细胞参照样本的毛细管电泳结果分析细胞遗传学异常;当所述细胞检测样本的毛细管电泳结果在所述细胞参照样本的毛细管电泳结果范围内,认为细胞遗传学正常,否则认为细胞遗传学异常。
35.在本发明中,分别分离细胞检测样本和细胞参照样本中的单个核细胞,制备为细胞悬液。在本发明中,将细胞检测样本与淋巴细胞分离液混合、离心;所述细胞检测样本与淋巴细胞分离液的体积比优选为1:1~2:1,更优选为1:1。在本发明中,所述离心的转速优选为1800rpm,所述离心的时间优选为20min;所述离心的温度优选为20℃,在本发明中,所述离心优选的采用高速冷冻离心机(thermofisher st 16r)设备进行;所述离心以“1”梯度缓升缓降。本发明在所述离心后,所述细胞检测样本和细胞参照样本分为4层,自上而下依次为血浆层、分离液层、粒细胞层和红细胞层;所述单个核细胞处于血浆层和分离液层中间。本发明收集所述单个核细胞,与pbs混合后,离心清洗,制备成细胞悬液。本发明对所述细胞悬液的浓度没有特殊限定。
36.本发明在获得所述细胞悬液后,将所述细胞悬液与cd138磁珠混合分选获得cd138阳性浆细胞。在本发明中,所述细胞悬液与cd138磁珠的体积比优选为10:1;所述混合的温度优选为3~5℃,更优选为4℃;所述混合的时间优选为25~35min,更优选为30min。本发明在所述混合后,过ms柱子;然后清洗所述ms柱子,所述清洗的次数优选为2~4次,更优选为3次;所述清洗优选的采用磁珠分选缓冲液进行;本发明在所述清洗后,将所述ms柱子移出磁场后,进行冲洗,收集洗脱液获得cd138阳性浆细胞。
37.本发明在获得所述cd138阳性浆细胞后,提取所述cd138阳性浆细胞的基因组dna。本发明对所述基因组dna的提取方法没有特殊限定,采用本领域常规的基因组dna提取方法即可。在本发明中,由于mlpa实验对dna样品中的污染物特别是盐离子的敏感性很强,所以所述基因组dna的a260/a280优选为1.6~1.9之间;所述基因组dna的浓度优选为20~30ng/μl;当所述基因组浓度大于30ng/μl时,优选的进行稀释。在本发明中,所述基因组dna优选的用te缓冲液溶解,不能用去离子水代替,以此避免dna高温时脱嘌呤的现象。在本发明中,
所述细胞检测样本和细胞参照样本优选的来源一致,提取方法一致;以避免因组织间拷贝数差异或dna提取方法间的差异带来的实验误差。在本发明中,所述细胞检测样本和细胞参照样本优选的避免使用肝素抗凝;本发明在提取dna时优选的避免使用qiagen ez1,m6,m48和m96试剂盒,以免在dna中残留较多的盐,影响反应。在本发明中,所述细胞检测样本和细胞参照样本的数量比为(7~15):1。在本发明具体实施过程中,所述细胞参照样本的数量优选的≥3。在本发明中,当细胞检测样本超过21个后,每增加7个标本,增加1个细胞参照样本。在本发明中,优选的设置空白对照样本,所述空白对照样本以h2o代替基因组dna。
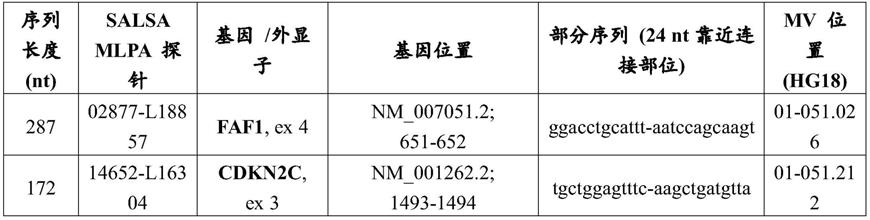
38.本发明在获得所述基因组dna后,将所述基因组dna变性,与mlpabuffer和mplaprobe mix混合进行mlpa反应获得mlpa产物。在本发明中,所述变性的的程序优选为98℃,5min变性;25℃,10min;在本发明中,所述变性后的基因组dna、mlpa buffer和mplaprobe mix的体积比优选为5:1.5:1.5。在本发明中,所述基因组dna、mlpabuffer和mplaprobe mix优选的置于pcr反应管中,每一个pcr反应管中,所述基因组dna的体积优选为5μl,所述mlpabuffer和mplaprobe mix的体积分别优选为1.5μl。在本发明中,所述mlpa反应的程序优选为95℃,1min;60℃,16~20h。在本发明中,所述mplaprobe mix包括核苷酸序列如seq id no.1~seq id no.53所示的探针,具体见表1;所述mlpabuffer属于高盐溶液,因此即使存储在-20℃也能保持液态。
39.本发明在获得所述mlpa产物后;将所述mlpa产物与连接酶混合液混合进行连接获得连接产物。在本发明中,所述连接酶混合液优选的包括连接酶缓冲液和连接酶。在本发明中,所述mlpa产物与连接酶混合液的体积比优选为1:4。在本发明中,所述mlpa产物优选的54℃,保持10min后;进行连接,所述连接的程序优选的为54℃,15min;98℃,5min;20℃保持。
40.在本发明中,所述连接酶缓冲液中含有甘油,因此在-20℃保持时,仍能保持液态。本发明中,所述连接酶混合液不能振荡,连接酶最添加;在加酶之后优选的用移液枪吹打混匀。如果连接酶与连接酶缓冲液没有充分混匀,可能会影响结果的准确性;如果混匀时过于激烈,可能导致酶失活。在本发明中,所述连接酶混合液优选的在使用之前一个小时内配好,并置于冰上备用。
41.本发明在获得所述连接产物后,将所述连接产物和pcr聚合酶混合液混合进行pcr扩增获得pcr扩增产物。在本发明中,所述的连接产物和pcr聚合酶混合液的体积比优选为4:1;所述pcr扩增的程序如下:95℃、30s;60℃、30s,72℃、60s,35个循环;72℃、20min;15℃停止。在本发明中,所述pcr聚合酶混合液优选的包括h2o、salsapcr-primer mix和salsapolymerase;在本发明中,所述h2o、salsapcr-primer mix和salsa polymerase的体积比优选为7.5:2:0.5;在本发明中,所述salsa pcr-primer mix包括pcr扩增引物。
42.本发明对所述pcr扩增产物进行毛细管电泳,比较细胞检测样本和细胞参照样本的毛细管电泳结果分析细胞遗传学异常;当所述细胞检测样本的毛细管电泳结果在所述细胞参照样本的毛细管电泳结果范围内,认为细胞遗传学正常,否则认为细胞遗传学异常。在本发明中,所述毛细管电泳优选的采用abi 3130xl仪器进行,所述毛细管电泳的染料优选为fam,所述毛细管的规格优选为36或50cm;所述毛细管电泳的注射混合物优选的包括pcr扩增产物、rox(或者liz)和甲酰胺;在本发明中,所述pcr扩增产物、rox(或者liz)和甲酰胺的体积比优选为0.7:0.3:9或0.7:0.2:9。在本发明中,所述毛细管电泳的参数优选的设置
如下:注射电压:1.6kv;注射时间:15s;运行电压:15kv或10kv;多聚物:pop4(15kv)或pop7(10kv);运行时间30min或40min。本发明在所述毛细管电泳前,优选的密封注射所述注射混合物,80℃变性2min,迅速冷却。
43.在本发明所述应用mlpa检测细胞遗传学异常的方法中,对所述检测样本没有特殊限定,任何细胞样本均可,在本发明中,以骨髓样本为例。本发明所述的应用mlpa检测细胞遗传学异常的方法为非诊断目的的方法,所述方法意在检测细胞学异常,并不用于检测疾病或预后评估。
44.本发明还提供了应用mlpa检测细胞遗传学异常的试剂盒,包括所述的mplaprobe mix。在本发明中,所述试剂盒优选的还包括所述的pcr聚合酶混合液;所述pcr聚合酶混合液包括pcr扩增引物。在本发明中,所述试剂盒优选的还包括上述检测方法中用到的试剂。
45.下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
46.实施例1
47.样品制备
48.1)提取待检测样本中单个核细胞:取淋巴细胞分离液ficol于离心管,将新鲜骨髓悬液沿管壁缓慢加入,离心1800rpm,20min,20℃,高速冷冻离心机(thermofisher st16r)以一级梯度缓升缓降。离心后可见试管内的血液清楚地分为4层,上层为血浆层,中层为分离液层,单个核细胞所处于血浆层和分离液层中间,底层为红细胞层,红细胞层上为粒细胞层;收集单个核细胞。后用pbs离心清洗,制成1
×
108个细胞/ml细胞悬液,备用。
49.2)分选浆细胞:取100μl细胞悬液加入10μl cd138磁珠,4℃孵育30min后过柱,用500μl的磁珠分选缓冲液(美天旎,货号130-091-221)冲洗柱子,(每次液体无残留时再加入新的液体)共三次,将柱子移开磁场到1.5ml ep管中,用1ml缓冲液稍用力冲洗,得到所需的cd138阳性浆细胞,流式检测阳性率>90%。
50.3)提取样本dna,并测od值。因mlpa实验对dna样品对污染物特别是盐离子的敏感性很强,所以应保证dna样品的质量,a260/a280应在1.6~1.9之间。注意事项:应用te溶解或稀释dna,不可用去离子水替代,以免dna高温时脱嘌呤;应保证所有参照样本及待检测样本组织来源的一致性及提取方法的一致性,以避免因组织间拷贝数差异或dna提取方法间的差异带来的实验误差;样本避免使用肝素抗凝;提取dna时避免使用qiagen ez1,m6,m48和m96试剂盒,以免在dna中残留较多的盐,影响反应;每次mlpa实验均需要使用参考样本dna进行对照。每次实验最少要设置3个参考样本dna对照。当检测标本超过21个,每增加7个标本,增加1个参考样本dna对照。放置参考样本的pcr管应该随机放置;空白对照:空白对照中dna被h2o取代;稀释或浓缩dna到合适浓度:dna浓度在10~50ng/μl之间,总量在50~250ng之间;实验前应对不同dna溶液进行一定的稀释使其浓度差不多在同一水平(优选是20~30ng/μl)。
51.dna变性+杂交
52.1)每个0.2ml的pcr管,做好标记,加5μl dna(20ng/μl);每反应的样本体积不要超过5μl,否则将会降低探针和盐浓度,影响探针与样本结合的速度和稳定性。
53.2)把pcr管置于普通杂交仪中开始mlp程序:
54.98℃,5min变性(体积5μl);然后25℃保持10min,加杂交混合液。
55.3)配制杂交缓冲液:mlpabuffer和mplaprobe mix在使用之前需要振荡混匀(经过冰冻-溶解过程后,盐类和其他成分可能会沉积到试管底部);mlpa缓冲液属于高盐溶液,因此即使存储在-20℃也能保持液态。
56.每个反应需要包含以下成分:1.5μl mlpabuffer+1.5μl probe mix,用枪吹打或者振荡混匀。可以多配5~10%的量以防移液所造成的损失。
57.4)当温度停在25℃时,将pcr管从杂交仪中取出。每个pcr管中加入3μl杂缓冲液,用枪慢慢吹打混匀(或者离心一下)。
58.继续程序:95℃
×
1min(体积8μl),然后60℃,16~20h复性。
59.表1探针的核苷酸序列
60.61.62.[0063][0064]
连接反应
[0065]
1)配制连接酶缓冲液:ligase buffer在使用以前要进行振荡混匀;连接酶缓冲液:25μl dh2o+3μl ligase-65buffera+3μl ligase-65buffer b,用枪吹打或者振荡混匀。
[0066]
2)加入连接酶:向连接酶缓冲液中加入1μl ligase-65,总体积为32μl。可以多配5~10%的量以防移液所造成的损失。
[0067]
3)继续程序:54℃保持10min,向每个pcr管中加入32μl连接酶混合液(总体积达到40μl),用移液枪慢慢吹打混匀。然后54℃,15min;98℃,5min灭活连接酶,最后20℃保持,此时可以将pcr管从杂交仪中取出。
[0068]
连接反应产物可以在20℃存放数小时,在4℃存放数周。
[0069]
pcr反应
[0070]
1)振荡混匀salsapcrprimer mix;用手握住polymerase 10s以降低粘性;
[0071]
2)配制聚合酶混合液(polymerase master mix):每个反应应该包括7.5μl dh2o+
2μl salsapcr-primer mix和0.5μl salsapolymerase。总体积10μl应该多配5~10%的量以防移液所造成的损失。用枪慢慢吹打混匀,不可振荡,应该在使用之前一个小时内配好,并置于冰上。
[0072]
3)室温下,向每一个pcr管中加入10μl聚合酶混合液。用枪慢慢吹打混匀,不可振荡。
[0073]
继续反应程序。
[0074]
95℃
×
30s(体积50μl);60℃
×
30s,72℃
×
60s,35个循环;72℃
×
20min;15℃暂停
[0075]
3)立即进行毛细管电泳,或者将pcr产物储存。4℃能够储存一周,-15~-25℃可以储存更长时间。pcr产物应该避光保存。
[0076]
毛细管电泳
[0077]
所用仪器:abi 3130xl
[0078]
染料:fam;
[0079]
推荐毛细管:36cm.
[0080]
注射混合物:0.7μl pcr扩增产物+0.3μl rox+9μl甲酰胺
[0081]
毛细管仪器设置:注射电压:1.6kv;注射时间:15s;运行电压:15kv;多聚物:pop4;运行时间30min。
[0082]
方法:密封注射后混合物,80℃变性2min,迅速冷却。
[0083]
结果分析:
[0084]
本发明利用上述方法对121例骨髓瘤患者细胞遗传学异常进行分析,其中表2为探针的参考范围,表3为细胞遗传学异常检出率。
[0085]
表2通过9个正常人的mlpa检测结果建立每个探针的参考范围
[0086]
[0087]
[0088][0089]
表3 mlpa检测121例骨髓瘤患者细胞遗传学异常检出率
[0090]
[0091][0092][0093]
表4 mlpa检测骨髓瘤细胞遗传学异常与fish检测对比
[0094][0095]
表5 mlpa和fish的敏感度比较
[0096][0097]
表4和表5为mlpa法与目前金标准fish检测骨髓瘤细胞遗传学异常结果对比和敏感度比较,由此可见,mlpa法检测骨髓瘤细胞遗传学异常的敏感度和特异度均较高,且对于13q缺失,mlpa技术可检测到小于20kb的dna缺失,且不仅限于发现特异性探针结合的13q异常。在保证检出率和准确度的同时,mlpa探针混合体系可以同时检测多达100个基因区域的异常,而fish值可以检测少数特异性探针区域,且快速、简单、高通量、重复性高,对mm的细胞遗传学检测有临床推广潜力。
[0098]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 用于检测AEDs引起的SJS/TEN的引物、试剂盒及方法与制造工艺
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺
- 用于多重PCR检测致腹泻寄生虫的引物组及试剂盒的制造方法与工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种基于特异性SS-COI引物的丽草蛉PCR快速检测方法与制造工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 紫苑石斛的分子特异性标记引物及检测方法
- 一种检测牛hcd携带者的特异性pcr引物、试剂盒及检测方法