五元杂环并苯环类化合物及其制备方法和医药用途与流程
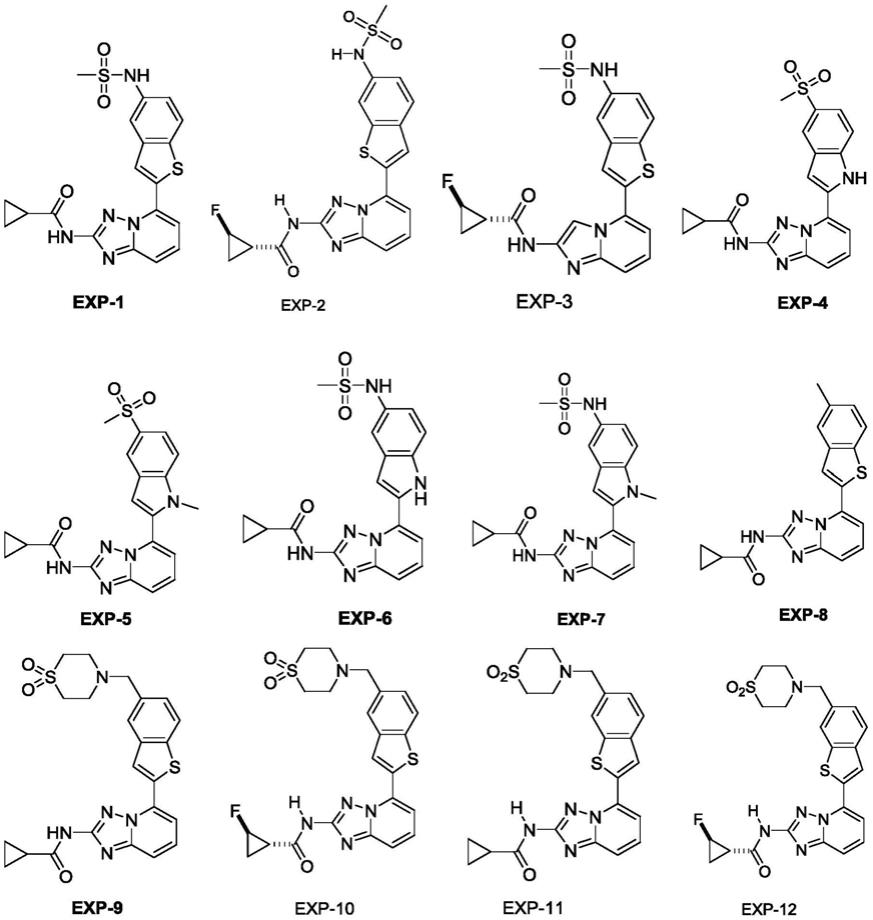
1.本发明属于医药技术领域,具体涉及一种新颖的五元杂环并苯环类化合物及其制备方法和应用。
背景技术:
2.软骨细胞是无血管组织的主要细胞组分,正常关节软骨中软骨细胞占组织体积的约5%,而细胞外基质占组织剩余的95%。软骨细胞分泌基质组分,主要是蛋白聚糖和胶原,其反过来为软骨细胞在机械压力下提供适合于它们生存的环境。在软骨中,ii型胶原与ix型蛋白胶原形成纤维样结构,为软骨提供很大的机械强度。蛋白聚糖可以吸收水并且负责软骨的弹性和减震性质。软骨的功能性作用之一是为骨头部分提供彼此光滑的关节连接。因此,关节软骨的损耗引起骨彼此摩擦,导致疼痛和运动丧失。在炎性关节炎例如类风湿性关节炎中,软骨退化是由发炎组织(例如发炎的滑膜)分泌的蛋白酶(胶原酶)引起的。受损软骨中的软骨细胞常常显示出软骨合成活性的减弱和/或软骨退化活性的增强,软骨变性是发生类风湿性关节炎和骨关节炎的主要疾病标志。类风湿性关节炎(ra)是慢性关节变性疾病,其特征在于关节结构的炎症和破坏,可能导致关节功能性的失常引起实质性的失能和疼痛,甚至过早死亡。因此,ra治疗的目的不仅是延缓疾病,减轻病痛,保护关节破坏。
3.jak激酶属于细胞质酪氨酸激酶的janus激酶(jak)家族,其参与细胞因子受体介导的细胞内信号转导。jak激酶家族包括4个成员:jak1、jak2、jak3和tyk2。jak募集细胞因子受体,结合细胞因子,随后将细胞因子受体和共享的受体亚基进行二聚化。然后jak通过自磷酸化和/或另一个jak的转磷酸化而活化,导致受体磷酸化以及信号转导物和转录激活物(stat)成员的募集和磷酸化。磷酸化的sata二聚化并且转移至细胞核内,在细胞核内它们结合至细胞因子应答基因的增强子区。jak在调控多种细胞因子受体家族的生物学应答功能中起着重要的作用。jak1基因敲除的小鼠具有早期的出生后致死因子显型,神经系统也受到损害,导致幼鼠出现先天缺陷。研究表明jak1基因敲除小鼠会出现胸腺细胞和b细胞的分泌缺陷,jak1基因敲除的组织对il-6、il-10的反应明显减弱。
4.临床已经确定jak与自身免疫疾病的关联,约70%的重症联合免疫缺陷病例中检测到jak以及上游信号传导组分r-c受体链和il7受体的突变。
5.临床上jak家族成员jak2涉及的病症,包括骨髓增殖性障碍、癌症特别是白血病例如急性髓性白血病、急性淋巴细胞白血病或实体瘤例如子宫平滑肌肉瘤、前列腺癌等相关疾病。
技术实现要素:
6.本发明提供新颖的五元杂环并苯环化合物的制备方法和医药用途。
7.更具体而言,本发明涉及结构如式(i)所示的化合物或其立体异构体、互变异构体或其药学上可接受的盐或其溶剂合物或者前药:
[0008][0009]
其中:
[0010]
a独立选自s、n、n(me);
[0011]
b独立选自取代或未取代的c
3-c6环烷基、具有1至2个独立选自n、o和s的环杂原子的5至6元杂芳基环,其中所述的5至6元杂芳环任选地被独立选自卤素、氟代或未氟代的c
1-c6烷基、氟代或未氟代的c
1-c6烷氧基、氟代或未氟代的c
1-c6烷基胺基的一个或多个取代基取代;
[0012]
d独立选自c,n;
[0013]
l1不存在或独立选自单键,-ch
2-,-(ch2)
x
o(ch2)
y-,-(ch2)
x
nh(ch2)
y-,-ch2o-,-c(o)-,-con(r4)-,-ch2n(r4)-,-conh(ch2)
y-,-n(r4)-,-so2n(r4)-,-s(o)
2-,-n(me)-;
[0014]
r4独立选自h、c
1-c6烷基、取代的c
1-c6烷基、c
1-c3醚c
1-c3烷基、甲磺酰基;
[0015]
r1独立选自h、-nh2、-cooh、取代或未取代的c
1-c6烷基、酰基、取代的或未取代的酰基胺基、取代的或未取代的c
1-c6烷氧基、卤素、羟基、取代的或未取代的c
1-c3酯基、取代或未取代的杂芳基;
[0016]
r3独立地选自h、取代或未取代的以下基团:磺酰基、磺酰胺基、5至6元杂环,其具有至少一个杂原子且任选地具有独立选自n和s的第二环杂原子、取代的或未取代的c
1-c6烷烃,取代基独立选自5至6元杂芳基和甲氧基取代、取代的或未取代的c
3-c7环烷基、取代的或未取代的4-7元杂环烷基、取代的或未取代的5-7元杂芳基。
[0017]
m是0、1、2、3;n是0,1、2、3;t是0、1、2、3。
[0018]
在一种实施方式中,其特征在于b选自取代或未取代的以下基团:环丙烷、环丁烷、环戊烷、环己烷、吡唑基、吡啶基、噻吩、噻唑、1,3,4-噻二唑、噁唑、1,3,4-噁二唑、呋喃、吡咯、3-吡咯啉、2-吡唑啉、咪唑、1,2,3-三唑、1,2,4-三唑、吡喃、哒嗪、嘧啶、吡嗪;取代基选自卤素、c
1-c3烷基、c
1-c3卤代烷基;优选的,b选自取代或未取代的以下基团:环丙烷、环丁烷、吡唑基、吡啶基、咪唑基;取代基选自f、cl、br、甲基、乙基、丙基、异丙基、三氟甲基。
[0019]
在一种实施方式中,d为c或n。
[0020]
在一种实施方式中,n为1。
[0021]
在一种实施方式中,r1独立选自h、-nh2、-cooh、羟基、卤素、取代或未取代的c
1-c3烷基、取代或未取代的c
1-c3酰基、取代的或未取代的c
1-c3酰基胺基、取代的或未取代的c
1-c3烷氧基、取代的或未取代的c
1-c3酯基;在一种优选的实施方式中,r1独立选自h、-nh2、-cooh、羟基、卤素、甲基、乙基、丙基、异丙基、甲酰基胺基、甲酯基。
[0022]
在一种实施方式中,m为0或者1,更优选的,m为0。
[0023]
在一种实施方式中,l1不存在或独立选自单键,-ch
2-,-(ch2)
x
o(ch2)
y-,-(ch2)
x
nh(ch2)
y-,-conh(ch2)
y-,-n(r4)-;r4独立选自h、甲基、乙基、丙基、乙基甲醚、甲基甲醚、乙基
乙醚;x或y独立的选自0、1或2。在一种更优选的实施方式中,l1不存在或独立选自单键,-ch
2-,-(ch2)
x
o(ch2)
y-,-(ch2)
x
nh(ch2)
y-,-conh(ch2)
y-,-n(r4)-;r4独立选自h、甲基、乙基甲醚;x或y独立的选自0、1或2。
[0024]
在一种实施方式中,t为1。
[0025]
在一种实施方式中,r3独立地选自h、取代或未取代的以下基团:磺酰基、磺酰胺基、硫代吗啉1,1-二氧化物、哌啶、吡唑、噻唑、咪唑、1,3,4-噻二唑、哌嗪、吗啉、嘧啶、吡嗪、噻吩、噁唑、1,3,4-噁二唑、呋喃、吡咯、3-吡咯啉、2-吡唑啉、1,2,3-唑、1,2,3-三唑、1,2,4-三唑、吡喃;取代基选自h、卤素、c
1-c3烷基、c
1-c3卤代烷基、羟基c
1-c3烷基、c
3-c6环烷基、乙基甲醚、甲基甲醚、乙基乙醚;优选的,r3独立地选自h、取代或未取代的以下基团:磺酰基、磺酰胺基、硫代吗啉1,1-二氧化物、哌啶、吡唑、噻唑、咪唑、1,3,4-噻二唑、哌嗪、吗啉、嘧啶、噻吩、噁唑、1,3,4-噁二唑、呋喃、吡咯、3-吡咯啉、2-吡唑啉、1,2,3-唑、1,2,3-三唑、1,2,4-三唑;取代基选自h、卤素、甲基、乙基、丙基、异丙基、三氟甲基、羟乙基、羟甲基、环丙烷、环丁烷、乙基甲醚、甲基甲醚、乙基乙醚。
[0026]
本发明还提供以下结构所示的化合物或其立体异构体、互变异构体或其药学上可接受的盐或其溶剂合物或者前药:
[0027]
[0028]
[0029]
[0030][0031]
本发明还提供了合成本发明化合物的方法以及公开的代表性的合成方案和路线。
[0032]
本发明还提供一种药物组合物,其包含本发明所述的式i的化合物或其药学上可接受的盐,和药学上可接受的稀释剂或载体。
[0033]
本发明还提供本发明式i的化合物在用于制备预防或治疗jak激酶相关疾病药物中的用途,特别是在制备用于预防和/或治疗涉及软骨退化、骨和/或关节退化的疾病、涉及炎症或免疫响应的病症、内毒素驱动的疾病状态、癌症和器官移植排斥的药物中的应用。
[0034]
本发明还提供一种通过施用本发明所述化合物预防和/或治疗涉及软骨退化、骨和/或关节退化的疾病、涉及炎症或免疫响应的病症、内毒素驱动的疾病状态、癌症和器官移植排斥的方法。
[0035]
在一种实施例中,本发明所述的涉及软骨退化、骨和/或关节退化的疾病、涉及炎症或免疫响应的病症、内毒素驱动的疾病状态、癌症和器官移植排斥的实例包括但不限于:骨关节炎、克隆病、类风湿性关节炎、银屑病、过敏性气道疾病(例如哮喘、鼻炎)、幼年特发性关节炎、结肠炎、炎性肠病、内霉素驱动的疾病状态、软骨更新损伤的疾病(例如软骨细胞的合成代谢兴奋的疾病)、先天软骨畸形、器官移植排斥、涉及软骨退化、关节退化、骨髓增
殖性障碍、白血病(急性髓性白血病、急性淋巴细胞白血病)、实体瘤(子宫平滑肌肉瘤、前列腺癌)等。
具体实施方式
[0036]
以下结合实施例进一步描述本发明,但这些实施例并非限制着本发明的范围,凡在本发明的构思前提下对本发明制备方法的简单改进都属于本发明的保护范围之内。下面实施例未注明具体条件的实验方法,通常按照本领域的公知手段。下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
[0037]
实施例1:化合物exp-1的制备
[0038]
合成路线如下:
[0039][0040]
步骤1.1化合物1的制备
[0041]
5-硝基苯并噻吩(sm1)(100mg,0.558mmol)溶于乙醇(3ml)中,氩气氛围下向其中加入钯碳(5mg,纯度10%)。反应液用氢气置换几次并加热到40℃在30psi压力下持续搅拌16小时。反应液过滤,滤液真空浓缩得到黄色固体5-氨基苯并噻吩(1)(100mg,收率100%)。
[0042]
1h nmr(400mhz,cdcl3):δ=7.65(d,j=8.4hz,1h),7.39(d,j=5.6hz,1h),7.16(d,j=5.2hz,1h),7.12(d,j=2.4hz,1h),6.80(dd,j=8.4and2.4 hz,1h),3.62(brs,2h).
[0043]
步骤1.2化合物2的制备
[0044]
5-氨基苯并噻吩(1)(100mg,0.67mmol)和三乙胺(136mg,1.34mmol)溶于二氯甲烷(2ml)中并冷却到0℃。甲烷磺酰氯(92mg,0.804mmol)缓慢加入并在0-20℃下搅拌2小时。反应液用乙酸乙酯(10ml)和水(10ml)稀释分层,水相用乙酸乙酯(10ml*3)萃取。有机相用无水硫酸钠干燥过滤,滤液旋干得到粗品。粗品用硅胶柱层析(洗脱剂:0%~30%乙酸乙酯/石油醚)得到白色固体n-[苯并噻吩-5-基]-n-甲基磺酰甲磺酰胺(2)(100mg,收率48.86%)。
[0045]
1h nmr(400mhz,cdcl3):δ=7.97(d,j=8.4hz,1h),7.84(d,j=2.0hz,1h),7.58(d,j=5.2hz,1h),7.40(d,j=5.2hz,1h),7.31(dd,j=8.4and2.0 hz,1h),3.46(s,6h).
[0046]
步骤1.3化合物3的制备
[0047]
n-[苯并噻吩-5-基]-n-甲基磺酰甲磺酰胺(2)(50mg,0.164mmol)溶于四氢呋喃(0.5ml)中并降温至-75℃。正丁基锂(2.5m正己烷溶液,0.16ml)在-75℃氮气氛围下缓慢加入。搅拌60分钟后,硼酸三异丙氧酯(46mg,245.59umol)加入到反应液中并在-75℃下搅拌15分钟。缓慢升温到15-20℃且继续搅拌16小时。反应液用10ml 10%的盐酸溶液淬灭并用
乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥过滤,滤液旋干得到棕色油状固体[5-(甲磺酰胺)苯并噻吩-2-基]硼酸(3)(30mg,粗品),无需纯化直接用于下一步。
[0048]
步骤1.4化合物exp-1的制备
[0049]
[5-(甲磺酰胺)苯并噻吩-2-基]硼酸(3)(20mg,0.074mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(21mg,0.074mmol)溶于二氧六环(0.5ml)和水(0.1ml)中,在氮气氛围中快速加入碳酸钾(31mg,0.221mmol)和1,1-双(二苯基磷)二茂铁氯化钯(10.8mg,0.0148mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(10ml)和水(10ml)稀释,水相用乙酸乙酯(10ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩到的粗品用制备hplc纯化(分离柱:bostonprime c18 150*30mm*5μm;流动相:a:水(0.04%nh3h2o+10mm nh4hco3)和b:乙腈;梯度:b 25-55%)纯化,并得到白色目标产物n-[5-[5-(甲磺酰胺)苯并噻吩-2-基][1,2,4]三唑[1,5-a]吡啶-2-基]环丙烷甲酰胺(exp-1)(3.7mg,收率11%yield)。
[0050]
lcms:tr=1.860min in 10-80cd_7min_220&254_agilent.met,chromatography(xbrige c18 30*2.1mm*3.5μm),ms(esi)m/z=428.1[m+h]
+
.
[0051]
hplc:tr=2.06min in 10-80cd_8min.met,chromatography(xbridge c18 2.1*50mm*5μm).
[0052]
1h nmr(400mhz,dmso-d6):δ=11.25(s,1h),9.87(s,1h),8.81(s,1h),8.05(d,j=8.8hz,1h),7.86-7.65(m,4h),7.34(d,j=8.8hz,1h),3.02(s,3h),2.03-1.94(m,1h),0.91-0.84(m,4h).
[0053]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表1的实施例化合物。
[0054]
【表1】
[0055]
[0056][0057]
实施例2:化合物exp-4的制备
[0058]
合成路线如下:
[0059][0060]
步骤2.1化合物4的制备
[0061]
4-甲磺酰苯胺(sm2)0(1g,5.84mmol)溶于乙醇(20ml)中并且加入碘(1.48g,5.84mmol)和硫酸银(2.00g,6.42mmol)。反应液在50度下搅拌2小时。反应液过滤并且将滤液旋干得到棕色固体2-碘-4-甲磺酰基-苯胺(740mg,收率43%)。
[0062]
lcms:tr=0.714min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(merck rp18e 25-3mm),ms(esi)m/z=297.8[m+h]
+
.
[0063]
步骤2.2化合物6的制备
[0064]
2-碘-4-甲磺酰基-苯胺(4)(250mg,1.11mmol),n-[5-乙炔基-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(5)(328mg,1.11mmol),碘化亚铜(42mg,0.221mmol),三乙胺(559mg,5.53mmol)和三苯基磷二氯化钯(77mg,0.11mmol)溶于四氢呋喃(6ml)中并且置换氮气3次后在60度下搅拌1小时。该反应混合物用二氯甲烷(50ml)和水(50ml)稀释,水相用二氯甲烷(50ml
×
3)萃取,有机相用饱和食盐水(100ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。粗品用硅胶柱层析(洗脱剂:80%~90%乙酸乙酯/石油醚)得到黄色固体n-[5-[2-(2-氨基-5-甲磺酰基-苯基)乙炔基]-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(6)(140mg,收率23%)。
[0065]
lcms:tr=0.691min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatograph y(merck rp18e 25-3mm),ms(esi)m/z=396.0[m+h]
+
.
[0066]
步骤2.3化合物exp-4的制备
[0067]
n-[5-[2-(2-氨基-5-甲磺酰基-苯基)乙炔基]-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(6)(110mg,0.278mmol)溶于n,n-二甲基甲酰胺(2ml)后加入叔丁醇钾(62mg,0.556mmol)在90度下搅拌2小时。反应混合物用乙酸乙酯(30ml)和水(25ml)稀释,水相用乙酸乙酯(20ml)萃取,有机相合并后用食盐水(50ml)洗一次,无水硫酸钠干燥后过滤浓缩。粗品用碱性制备分离纯化得到白色固体目标产物n-[5-(5-甲磺酰基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(exp-4)(2.4mg,收率2%)。
[0068]
lcms:tr=2.448min in 10-80ab_7min_220&254_agilent.met,chromatography
(xbrige c18,30*2.1mm*3.5μm),ms(esi)m/z=396.1[m+h]
+
.
[0069]
hplc:tr=2.53min in 10-80cd_8min.met,chromatography(xbridge c18,2.1*50mm*5μm).
[0070]
1h nmr(400mhz,dmso-d6):δ=12.50(s,1h),11.28(s,1h),8.32(d,j=1.6h z,1h),8.24(s,1h),7.90-7.80(m,2h),7.79-7.69(m,3h),3.23(s,3h),2.13-2.05(m,1h),0.92-0.86(m,4h).
[0071]
实施例3:化合物exp-5的制备
[0072]
合成路线如下:
[0073][0074]
步骤3.1化合物exp-5的制备
[0075]
n-[5-(5-甲磺酰基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(exp-4)(30mg,0.076mmol)溶于n,n-二甲基甲酰胺(0.5ml)后加入碳酸钾(31mg,0.228mmol)和碘甲烷(11mg,0.083mmol)在10-12度下搅拌4小时。反应混合物用乙酸乙酯(20ml)和水(10ml)萃取,有机相用食盐水(20ml)洗一次,无水硫酸钠干燥后过滤浓缩。得到的粗品用碱性制备分离纯化得到白色固体目标产物n-[5-(1-甲基-5-甲磺酰基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(exp-5)(2.0mg,6%yield)。
[0076]
lcms:tr=2.246min in 10-80ab_7min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*30mm3um),ms(esi)m/z=410.0[m+h]
+
.
[0077]
hplc:tr=3.13min in 10-80ab_8min.met,chromatography(ultimate c18 3.0*50mm3um).
[0078]
1h nmr(400mhz,dmso-d6):δ=11.13(s,1h),8.29(d,j=1.2hz,1h),7.91-7.83(m,2h),7.82-7.75(m,2h),7.39(dd,j=7.2,0.8hz,1h),7.15(s,1h),3.74(s,3h),3.20(s,3h),2.04-1.91(m,1h),0.90-0.68(m,4h).
[0079]
实施例4:化合物exp-6的制备
[0080]
合成路线如下:
[0081][0082]
步骤4.1化合物8的制备
[0083]
n-[5-(5-硝基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺
(70mg,0.193mmol)溶于乙醇(1ml)中并且在氮气氛围下加入湿钯碳(7mg),该反应体系置换氢气3次并在30度(30psi)下搅拌4小时。反应混合物过滤并且将滤液旋干得到粗品。得到的粗品用硅胶柱层析(洗脱剂:5%甲醇/二氯甲烷)得到黄色固体n-[5-(5
–
氨基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(8)(25mg,收率32%)。
[0084]
lcms:tr=0.709min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(merck rp-18e 25-3mm),ms(esi)m/z=333.0[m+h]
+
.
[0085]
步骤4.2化合物exp-6的制备
[0086]
n-[5-(5
–
氨基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(8)(25mg,0.075mmol)溶于二氯甲烷(1ml)中并加入三乙胺(22mg,0.226mmol)和甲磺酰氯(9mg,0.083mmol)仍后在10-17度下搅拌1个小时。该反应液用饱和的碳酸氢钠(10ml)淬灭并且用二氯甲烷(10ml x 2)萃取。有机相合并用食盐水(10ml)洗一次,无水硫酸钠干燥后过滤浓缩。得到的粗品用碱性制备分离纯化得到白色固体目标产物n-[5-(5-甲磺酰胺基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(exp-6)(2mg,收率6%)。
[0087]
lcms:tr=2.353min in 10-80ab_7min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*30mm3um),ms(esi)m/z=411.0[m+h]
+
.
[0088]
hplc:tr=2.358min in 10-80cd_8min.met,chromatography(xbridge c18 2.1*50mm 5um).
[0089]
1h nmr(400mhz,dmso-d6):δ=11.96(s,1h),11.25(s,1h),9.31(s,1h),8.02(s,1h),7.79(d,j=5.2hz,2h),7.69-7.63(m,1h),7.55-7.44(m,2h),7.15(dd,j=8.8,2.0hz,1h),2.90(s,3h),2.12-1.95(m,1h),0.93-0.82(m,4h).
[0090]
实施例5:化合物exp-5的制备
[0091]
合成路线如下:
[0092][0093]
步骤5.1化合物9的制备
[0094]
n-[5-乙炔-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(5)(520mg,2.30mmol),2-碘-4-硝基-苯胺(sm3)(606mg,2.30mmol),碘化亚铜(87mg,0.46mmol),三乙胺(1.16g,11.49mmol)和三苯基磷二氯化钯(161mg,0.23mmol)溶于四氢呋喃(6ml)中并且置换氮气3次后在60度下搅拌1小时。向反应混合物加水(50ml)并且用二氯甲烷(50mlx2)萃取,有机相用饱和食盐水(50ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用
硅胶柱层析(洗脱剂:70%乙酸乙酯/石油醚)得到黄色固体n-[5-[2-(2-氨基-5-硝基-苯基)乙炔]-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(9)(340mg,36%yield)。
[0095]
lcms:tr=0.779min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(merck rp18e 25-3mm),ms(esi)m/z=363.0[m+h]
+
.
[0096]
步骤5.2化合物7的制备
[0097]
n-[5-[2-(2-氨基-5-硝基-苯基)乙炔]-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(9)(340mg,0.94mmol)溶于n,n-二甲基甲酰胺(5ml)中加入叔丁醇钾(210mg,1.88mmol)并且在90度下搅拌2小时。该反应混合物加入水(10ml)并且用乙酸乙酯(20ml)萃取,有机相用饱和食盐水(20ml)洗两次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:80%乙酸乙酯/石油醚)得到黄色固体n-[5-(5-硝基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(24)(200mg,58%yield)。
[0098]
1h nmr(400mhz,dmso-d6):δ=12.65(s,1h),11.26(s,1h),8.72(d,j=2.0hz,2h),8.25(s,1h),8.13(dd,j=9.2,2.4hz,1h),7.89-7.72(m,3h),7.69(d,j=9.2hz,1h),2.17-2.02(m,1h),1.01-0.76(m,4h).
[0099]
步骤5.3化合物10的制备
[0100]
n-[5-(5-硝基-1h-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(7)(190mg,0.524mmol)溶于n,n-二甲基甲酰胺(2ml)中加入碳酸钾(217mg,1.57mmol)和碘甲烷(81mg,0.577mmol)并在30度下搅拌2小时。该反应混合物加入水(10ml)并用乙酸乙酯(20ml)萃取,有机相用饱和食盐水(20ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:80%乙酸乙酯/石油醚)得到黄色固体n-[5-(1-甲基-5-硝基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(10)(90mg,32%yield)。
[0101]
lcms:tr=0.753min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(merck rp18e 25-3mm),ms(esi)m/z=377.0[m+h]
+
.
[0102]
1h nmr(400mhz,dmso-d6):δ=11.13(s,1h),8.71(d,j=2.4hz,1h),8.17(dd,j=9.2,2.0hz,1h),7.93-7.75(m,3h),7.41(d,j=7.2hz,1h),7.21(s,1h),3.75(s,3h),2.22-1.95(m,1h),0.83-0.60(m,4h).
[0103]
步骤5.4化合物11的制备
[0104]
n-[5-(1-甲基-5-硝基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(10)(80mg,0.212mmol)溶于乙醇(1ml)中在氮气的氛围中加入钯碳(10mg,钯纯度10wt%)并且置换氢气3次,该反应体系在氢气(30psi)氛围30度下搅拌4小时。反应液过滤后滤液旋干得到粗品,得到的粗品用硅胶柱层析(洗脱剂:4%甲醇/二氯甲烷)得到黄色固体n-[5-(1-甲基-5
–
氨基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(11)(30mg,34%yield)。
[0105]
lcms:tr=0.563min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(merck rp18e 25-3mm),ms(esi)m/z=347.1[m+h]
+
.
[0106]
步骤5.5化合物exp-7的制备
[0107]
n-[5-(1-甲基-5
–
氨基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(11)(30mg,0.087mmol)溶于二氯甲烷(1ml)中加入三乙胺(26mg,0.26mmol)和甲磺酰氯(10mg,0.095mmol)并且在10-17度下搅拌16小时。该反应液用饱和的碳酸氢钠(10ml)淬灭
并且用二氯甲烷(10ml x 2)萃取。有机相合并用食盐水(10ml)洗一次,无水硫酸钠干燥后过滤浓缩。得到的粗品用碱性制备分离纯化得到白色固体目标产物n-[5-(5-甲磺酰基-1-甲基-吲哚-2-基)-[1,2,4]三唑[1,5-a]并吡啶-2-基]环丙甲酰胺(exp-7)(2.3mg,6%yield)。
[0108]
lcms:tr=2.231min in 10-80ab_7min_220&254_agilent.met,chromatography(xtimate c18 2.1*30mm3um),ms(esi)m/z=447.0[m+na]
+
.
[0109]
hplc:tr=3.10min in 10-80cd_8min.met,chromatography(ultimate c18 3.0*50mm3um).
[0110]
1h nmr(400mhz,dmso-d6):δ=11.12(s,1h),9.32(s,1h),7.87-7.73(m,2h),7.63-7.50(m,2h),7.34(dd,j=7.2,1.6hz,1h),7.21(dd,j=8.8,2.0hz,1h),6.91(s,1h),3.66(s,3h),2.89(s,3h),2.05-1.90(m,1h),0.89-0.74(m,4h).
[0111]
实施例6:化合物exp-8的制备
[0112]
合成路线如下:
[0113][0114]
步骤6.1化合物12的制备
[0115]
5-甲基苯并噻吩(sm4)(200mg,1.35mmol)溶解在四氢呋喃(4ml)中,在-75℃氮气氛围下向其中缓慢滴加正丁基锂(2.5m正己烷溶液,0.81ml)。搅拌60分钟后,加入硼酸三异丙酯(381mg,2.03mmol),然后缓慢升到15-20℃并持续搅拌16小时。反应液用1m盐酸溶液(20ml)淬灭且水相用乙酸乙酯(20ml x 3)萃取,有机相用无水硫酸钠干燥,过滤,滤液旋干得到目标产物[5-甲基苯并噻吩-2-基]硼酸(29)(300mg,粗品,白色固体)。
[0116]
1h nmr(400mhz,dmso):δ=8.44(s,2h),7.87(s,1h),7.83(d,j=8.4hz,1h),7.68(s,1h),7.20(d,j=8.4hz,1h),2.41(s,3h).
[0117]
步骤6.2化合物exp-8的制备
[0118]
[5-甲基苯并噻吩-2-基]硼酸(12)(150mg,0.78mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(220mg,0.78mmol)溶于二氧六环(5ml)和水(1ml)中,在氮气氛围中快速加入碳酸钾(324mg,2.34mmol)和1,1-双(二苯基磷)二茂铁氯化钯(114mg,0.156mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用硅胶柱层析(洗脱剂:20%~100%乙酸乙酯/石油醚)得到粗品(180mg,纯度70%)。然后100mg粗品采用hplc纯化(分离柱:a gela asb 150*25mm*5um;流动相:a:水(0.05%hcl)和b:乙腈;梯度:b 50-80%)纯化,并得到黄色目标产物n-[5-[5-甲基苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-8)(14.3mg,收率4.64%,纯度97.53%,hcl盐),剩下的80mg粗品用于其他反应。
[0119]
lcms:tr=3.758min in 10-80cd_7min_220&254_agilent.met,chromatography(xbrige c18 30*2.1mm 3.5um),ms(esi)m/z=349.2[m+h]
+
.
[0120]
hplc:tr=3.61min in 10-80cd_8min.met,chromatography(xbridge c18 2.1*50mm*5um).
[0121]
1h nmr(400mhz,dmso-d6):δ=11.24(s,1h),8.77(s,1h),7.97(d,j=8.4hz,1h),7.81-7.70(m,4h),7.32(d,j=8.4hz,2h),2.46(s,3h),2.20-2.01(m,1h),0.92-0.82(m,4h).
[0122]
实施例7:化合物exp-9的制备
[0123]
合成路线如下:
[0124][0125]
步骤7.1化合物13的制备
[0126]
将[5-甲基苯并噻吩-2-基]硼酸(12)(170mg,0.89mmol)溶解在四氢呋喃(2ml)中,向其中加入嚬哪醇(104mg,0.89mmol)和三氟乙酸(20mg,0.18mmol)。反应液置换三次氮气后在15-20℃下反应2小时。反应混合物用乙酸乙酯(20ml)和水(20ml)稀释分层,水相用乙酸乙酯(20ml
×
3)萃取,有机相用食盐水(20ml
×
3)洗涤、无水硫酸钠干燥后过滤浓缩旋干后得到白色目标产物4,4,5,5-四甲基-2-(5-甲基苯并噻吩-2-基)-1,3,2-二噁硼烷(13)(200mg,收率82%),将其直接用于下一步的反应而无需进一步纯化。
[0127]
1h nmr(400mhz,dmso-d6):δ=7.89(d,j=8.4hz,1h),7.82(s,1h),7.74(s,1h),7.27(d,j=8.4hz,1h),2.42(s,3h),1.32(s,12h).
[0128]
步骤7.2化合物14的制备
[0129]
将4,4,5,5-四甲基-2-(5-甲基苯并噻吩-2-基)-1,3,2-二噁硼烷(13)(200mg,0.73mm ol)溶解在四氯化碳(5ml)中,向其中加入n-溴代丁二酰亚胺(130mg,0.73mmol)和过氧化苯甲酰(18mg,0.07mmol)后在80℃下持续搅拌2小时。反应混合物用乙酸乙酯(20ml)和水(20ml)稀释分层,水相用乙酸乙酯(20ml
×
3)萃取,有机相用食盐水(20ml
×
3)洗涤、无水硫酸钠干燥后过滤浓缩旋干后得到白色目标产物2-[5-(溴甲基)-苯并噻吩-2-基]-4,4,5,5-四甲基-1,3,2-二噁硼烷(14)(250mg,收率97%),将其直接用于下一步的反应而无需进一步纯化。
[0130]
1h nmr(400mhz,dmso-d6):δ=8.05-8.01(m,2h),7.92(s,1h),7.54-7.46(m,1h),4.86(s,2h),1.33(s,12h).
[0131]
步骤7.3化合物int-2的制备
[0132]
将2-[5-(溴甲基)-苯并噻吩-2-基]-4,4,5,5-四甲基-1,3,2-二噁硼烷(14)(250mg,0.71mmol)和碳酸钾(196mg,1.42mmol)溶解在乙腈(4ml)中,向其中加入1,1-二氧硫代吗啉(96mg,0.71mmol)后在15-20℃下持续搅拌2小时。反应液过滤旋干得到白色固体4-[[2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2基)苯并噻吩-5-基]甲基]-1,1-二氧硫代吗啉(int-2)(200mg,粗品),将其直接用于下一步的反应而无需进一步纯化。
[0133]
1h nmr(400mhz,dmso-d6):δ=7.98(d,j=8.4hz,1h),7.90-7.89(m,2h),7.42(d,j=8.4hz,1h),3.79(s,2h),3.15-3.08(m,4h),2.93-2.84(m,4h),1.32(s,12h).
[0134]
步骤7.4化合物exp-9的制备
[0135]
4-[[2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2基)苯并噻吩-5-基]甲基]-1,1-二氧硫代吗啉(int-2)(200mg,0.49mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(110mg,0.39mmol)溶于二氧六环(3ml)和水(0.6ml)中,在氮气氛围中快速加入碳酸钾(204mg,1.47mmol)和1,1-双(二苯基磷)二茂铁氯化钯(72mg,0.098mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用硅胶柱层析(洗脱剂:20%~100%乙酸乙酯/石油醚)得到粗品(180mg,纯度70%)。然后100mg粗品采用hplc纯化(分离柱:ageladurashell c18 150*25mm*5um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 35-65%)纯化,并得到白色目标产物n-[5-[5-[(1,1-二氧硫代吗啉-4-基)甲基]苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-9)(28.6mg,收率12%,纯度99.14%)。
[0136]
lcms:tr=3.420min in 10-80ab_7min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=482.3[m+h]
+
.
[0137]
hplc:tr=3.66min in 10-80ab_8min.met,chromatography.
[0138]
1h nmr(400mhz,dmso-d6):δ=11.23(s,1h),8.83(s,1h),8.06(d,j=8.4hz,1h),7.93(s,1h),7.79-7.70(m,3h),7.47(d,j=8.4hz,1h),3.83(s,2h),3.17-3.04(m,4h),3.01-2.89(m,4h),2.14-2.08(m,1h),0.97-0.78(m,4h).
[0139]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表2的实施例化合物。
[0140]
【表2】
[0141]
[0142][0143]
实施例8:化合物exp-14的制备
[0144]
合成路线如下:
[0145][0146]
步骤8.1化合物47的制备
[0147]
将6-溴-苯并噻吩(sm6)(100mg,0.47mmol)和甲烷磺酰胺(sm7)(67mg,0.70mmol)溶解在n,n-二甲基乙酰胺(2ml)中,室温下向其中加入碘化亚铜(9mg,0.47mmol)和碳酸钾(130mg,0.94mmol),然后在190℃下微波反应2小时。反应混合物过滤,滤液用乙酸乙酯(10ml)和水(10ml)稀释分层,水相用乙酸乙酯(10ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用硅胶柱层析(洗脱剂:0%~50%乙酸乙酯/石油醚)得到淡棕色目标产物n-(苯并噻吩-6-基)甲烷磺酰胺(47)(70mg,收率66%)。
[0148]
1h nmr(400mhz,dmso-d6):δ=9.83(s,1h),7.85-7.79(m,2h),7.67(d,j=5.2hz,1h),7.40(d,j=5.2hz,1h),7.25(dd,j=8.4,2.0hz,1h),3.00(s,3h).
[0149]
步骤8.2化合物48的制备
[0150]
n-(苯并噻吩-6-基)甲烷磺酰胺(47)(70mg,0.31mmol)溶解在四氢呋喃(2ml)中,在-75℃氮气氛围下向其中缓慢滴加正丁基锂(2.5m,0.62ml)。搅拌60分钟后,加入硼酸三异丙酯(174mg,0.92mmol),然后缓慢升到15-20℃并持续搅拌16小时。反应液用1m盐酸溶液(10ml)淬灭且水相用乙酸乙酯(10ml
×
3)萃取,有机相用无水硫酸钠干燥,过滤,滤液旋干得到淡蓝色固体[6-(甲烷磺酰胺)苯并噻吩-2-基]硼酸(48)(70mg,粗品)。
[0151]
1h nmr(400mhz,dmso-d6):δ=9.88(s,1h),8.46(s,2h),7.89(s,1h),7.84(d,j=8.8hz,1h),7.75(s,1h),7.23(dd,j=8.8,1.6hz,1h),3.01(s,3h).
[0152]
步骤8.3化合物exp-14的制备
[0153]
[6-(甲烷磺酰胺)苯并噻吩-2-基]硼酸(48)(70mg,0.26mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(58mg,0.21mmol)溶于二氧六环(1ml)和水(0.2ml)中,在氮气氛围中快速加入碳酸钾(107mg,0.77mmol)和1,1-双(二苯基磷)二茂铁氯化钯(19mg,0.026mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用hplc纯化(分离柱:ageladurashell c18 250*25mm*10um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 40-70%)纯化,并得到白色目标产物n-[5-[6-(甲烷磺酰胺)苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-14)(10.6mg,收率9%,纯度93.21%)。
[0154]
lcms:tr=3.880min in 10-80ab_7min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=428.2[m+h]
+
.
[0155]
hplc:tr=4.79min in 10-80ab_8min.met,chromatography.
[0156]
1h nmr(400mhz,dmso-d6):δ=11.21(s,1h),10.06(s,1h),8.78(s,1h),7.94(d,j=8.8hz,1h),7.86(s,1h),7.79-7.67(m,3h),7.32(dd,j=8.8,1.6hz,1h),3.08(s,3h),2.17-2.01(m,1h),0.97-0.75(m,4h).
[0157]
实施例9:化合物exp-15的制备
[0158]
合成路线如下:
[0159][0160]
步骤9.1化合物50的制备
[0161]
在0℃冰水浴条件下向(苯并噻吩-5-基)-氨基甲酸叔丁酯(49)(371mg,1.49mmol)的无水四氢呋喃(7.5ml)溶液中分批加入氢化钠(72mg,60%纯度于矿物油中,1.80mmol)。搅拌30分钟后加入碘甲烷(0.2ml,d=2.28g/ml,3.21mmol)。反应液逐渐升至室温并持续搅拌17小时。所得浅黄色浊液用20%氯化铵溶液(10ml)淬灭,再用叔丁基甲醚(12ml,10ml,8ml)萃取。合并有机相并用水(12ml
×
2)和饱和食盐水(15ml)洗涤,经无水硫酸钠干燥,过滤并减压浓缩后得到(苯并噻吩-5-基)-甲胺基甲酸叔丁酯(50)(366mg,1.39mmol,93%收率,浅黄色固体)并直接用于下一步反应。
[0162]1h nmr(400mhz,cdcl3):δ=7.80(d,j=8.4hz,1h),7.65(s,1h),7.46(d,j=5.2hz,1h),7.29(d,j=5.6hz,1h),7.23(dd,j=8.8,1.6hz,1h),3.31(s,3h),1.45(s,9h).
[0163]
步骤9.2化合物15的制备
[0164]
在-60℃氮气氛围下向(苯并噻吩-5-基)-甲胺基甲酸叔丁酯(50)(366mg,1.39mmol)的无水四氢呋喃(9.0ml)溶液中持续五分钟滴加正丁基锂(0.8ml,2.5m己烷溶液)。反应液维持在-60℃下继续搅拌50分钟后持续滴加硼酸三异丙酯(0.4ml,d=0.818g/ml)达五分钟,随后自然升至室温并持续搅拌17小时。所得黄色浊液用0.2m稀盐酸(10ml)淬灭后用乙酸乙酯(6ml
×
3)萃取。合并有机相后用饱和食盐水(10ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干得到5-(叔丁氧羰基)甲胺基苯并噻吩-2-硼酸(15)(443mg,约90%纯度,93%收率,浅黄色固体)并直接用于下一步反应。
[0165]1h nmr(400mhz,dmso-d6):δ=8.50(brs,2h),7.94-7.87(m,2h),7.77(d,j=1.0hz,1h),7.28(dd,j=8.8,2.0hz,1h),3.23(s,3h),1.38(s,9h).
[0166]
步骤9.3化合物16的制备
[0167]
向n-(5-溴-[1,2,4]三唑并[1,5-a]吡啶-2-基)-环丙甲酰胺(int-1)(244mg,0.868mmol)和5-(叔丁氧羰基)甲胺基苯并噻吩-2-硼酸(15)(443mg,90%纯度,1.30mmol)的1,4-二氧六环(7.0ml)和水(1.5ml)混合溶液中加入1,1
’-
双(二苯基膦)二茂铁二氯化钯(40mg,0.055mmol)和磷酸钾(503mg,2.37mmol)。该混合液用氮气置换数次后,在氮气氛围下加热至95℃并持续搅拌20小时。所得黄色悬浊液冷却后加水(15ml)稀释。过滤收集所生成的沉淀并用水(5ml
×
2)洗涤,再真空干燥得到{5-[5-(叔丁氧羰基)甲胺基苯并噻吩-2-基]-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(16)(495mg,约80%纯度,98%收率,浅棕色固体)并直接用于下一步反应。
[0168]
lcms:tr=0.998min in 5-95ab_1.5min_220&254_shimadzu.lcm,ms(esi)m/z=464.1[m+h]
+
.
[0169]1h nmr(400mhz,dmso-d6):δ=11.21(brs,1h),8.76(s,1h),8.04(d,j=8.8hz,1h),7.86(s,1h),7.81-7.68(m,3h),7.45-7.37(m,1h),3.27(s,3h),2.17-2.04(m,1h),1.41(s,9h),1.40-1.29(m,4h).
[0170]
步骤9.4化合物17的制备
[0171]
在10℃下将{5-[5-(叔丁氧羰基)甲胺基苯并噻吩-2-基]-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(16)(495mg,80%纯度,0.855mmol)悬浮于无水二氯甲烷(5.6ml)中并向其中滴加三氟乙酸(2.8ml,d=1.54g/ml,37.82mmol)。所得深棕色溶液在室温下持续搅拌2小时后,用氮气流鼓泡法除去大部分挥发物,再用1m碳酸钾溶液(20ml)处理。所得混合物用二氯甲烷/甲醇(30ml
×
3,v/v=5/1)混合萃取。合并萃取相用饱和食盐水(40ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干得到{5-(5-甲胺基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(17)(234mg粗品)并直接用于下一步反应。
[0172]
lcms:tr=0.783min in 5-95ab_1.5min_220&254_shimadzu.lcm,ms(esi)m/z=364.1[m+h]
+
.
[0173]
步骤9.5化合物exp-15的制备
[0174]
在10℃下向{5-(5-甲胺基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(17)(117mg)的无水二氯甲烷(4ml)悬浊液中加入三乙胺(0.1ml,d=0.727g/ml)和甲基磺酸酐(86mg)。反应液在10℃下持续搅拌20小时,将所得黄棕色悬浊液减压浓缩除去大部分挥发物后加水稀释。过滤收集生成的沉淀并用水(2ml
×
2)洗涤,再用高效液相色
谱(50-80%乙腈/0.04%氨水+10mm碳酸氢铵)制备分离,收集合并纯净组分,减压浓缩并冻干后得到最终产物{5-(5-甲烷磺酰甲胺基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(exp-15)(13.1mg,100%纯度,7%的两步收率,白色固体)。
[0175]
lcms:tr=2.907min in 10-80ab_4min_220&254_shimadzu.lcm,ms(esi)m/z=442.2[m+h]
+
.
[0176]
tr=2.975min in 10-80ab_4min_220&254_shimadzu.lcm,ms(esi)m/z=442.2[m+h]
+
.
[0177]
hplc:tr=2.64min in 10-80cd_8min.met(xbridge shield rp18 5um).
[0178]1h nmr(400mhz,dmso-d6):δ=11.24(brs,1h),8.83(s,1h),8.34-7.96(m,2h),7.95-7.36(m,4h),3.45(s,3h),3.00(s,3h),2.16-1.93(m,1h),1.10-0.60(m,4h).
[0179]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表3的实施例化合物。
[0180]
【表3】
[0181]
[0182]
[0183][0184]
实施例10:化合物exp-25的制备
[0185]
合成路线如下:
[0186][0187]
步骤10.1化合物exp-25的制备
[0188]
4-[[2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2基)苯并噻吩-5-基]甲基]吗啡啉(int-3)(100mg,0.28mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(39mg,0.14mmol)溶于二氧六环(0.5ml)和水(0.1ml)中,在氮气氛围中快速加入碳酸钾(115mg,0.83mmol)和1,1-双(二苯基磷)二茂铁氯化钯(20mg,0.027mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物和eb5-42合并过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤,浓缩,旋干后采用制备hplc纯化(分离柱:ageladurashell c18 250*25mm*10um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 45-75%)纯化,得到白色目标产物n-[5-[5-(吗啉)苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-25)(15mg,收率10%,纯度100%)。
[0189]
hplc:tr=2.75min in 10-80cd_8min.met,chromatography(xbridge shield rp18 5um).
[0190]
1h nmr(400mhz,dmso-d6):δ=11.22(s,1h),8.82(s,1h),8.04(d,j=8.4hz,1h),7.89(s,1h),7.80-7.68(m,3h),7.45(d,j=8.4hz,1h),3.66-3.54(m,6h),2.43-2.37(m,4h),2.14-2.02(m,1h),0.91-0.81(m,4h).
[0191]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表4的实施例化合物。
[0192]
【表4】
[0193][0194][0195]
实施例11:化合物exp-27的制备
[0196]
合成路线如下:
[0197][0198]
步骤11.1化合物int-4的制备
[0199]
将2-[5-(溴甲基)-苯并噻吩-2-基]-4,4,5,5-四甲基-1,3,2-二噁硼烷(14)(100mg,0.28mmol)和碳酸钾(78mg,0.57mmol)溶解在乙腈(2ml)中,向其中加入4,4-二氟哌啶(sm8)(34mg,0.28mmol)后在15-20℃下持续搅拌2小时。反应混合物用乙酸乙酯(20ml)和水(20ml)稀释分层,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后得到棕色目标产物4,4-二氟-1-[[2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2基)苯并噻吩-5-基]甲基]哌啶(int-4)(150mg,粗品),将其用于下面的反应而无需进一步纯化。
[0200]
步骤11.2化合物exp-27的制备
[0201]
4,4-二氟-1-[[2-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2基)苯并噻吩-5-基]甲基]哌啶(int-4)(100mg,0.25mmol)和(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺)环丙甲酰胺(int-1)(36mg,0.13mmol)溶于二氧六环(2ml)和水(0.4ml)中,在氮气氛围中快速加入碳酸钾(105mg,0.76mmol)和1,1-双(二苯基磷)二茂铁氯化钯(19mg,0.025mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后用制备hplc纯化(分离柱:ageladurashell c18 250*25mm*10um;流动相:a:水(0.04%nh
3.
h2o+10mm nh4hco3)和b:乙腈;梯度:b 55-85%)纯化,并得到淡黄色目标产物n-[5-[5-(4,4-二氟-1-哌啶)甲基]苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-27)(20mg,收率13%,纯度95.12%)。
[0202]
lcms:tr=3.324min in 10-80ab_7min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=468.3[m+h]
+
.
[0203]
hplc:tr=3.36min in 10-80cd_8min.met,chromatography(xbridge shield rp18 5um).
[0204]
1h nmr(400mhz,dmso-d6):δ=11.22(s,1h),8.82(s,1h),8.04(d,j=8.0hz,1h),7.89(s,1h),7.76-7.74(m,3h),7.45(d,j=8.4hz,1h),3.70(s,2h),3.31-3.27(m,4h),2.17-2.06(m,1h),2.04-1.87(m,4h),0.98-0.83(m,4h).
[0205]
实施例12:化合物exp-28、exp-29、exp-30、exp-31的制备
[0206]
合成路线如下:
[0207][0208]
步骤12.1化合物18的制备
[0209]
向5-溴苯并噻吩(sm9)(8.61g,39.6mmol)和氨基甲酸叔丁酯(6.45g,55.04mmol)的1,4-二氧六环(180ml)溶液中加入碳酸铯(25.67g,78.80mmol)和醋酸钯(569mg,2.53mmol),并在氮气流下加入4,5-双二苯基膦-9,9-二甲基氧杂蒽(2.66g,4.59mmol)。该混合液用氮气置换数次后,在氮气氛围下加热至100℃并持续搅拌20小时。所得深棕色悬浊液冷却后加乙酸乙酯(200ml)稀释后过滤除去不溶物。滤液依次用水(200ml)和饱和食盐水(200ml)洗涤,经无水硫酸钠干燥,过滤后减压浓缩。残留物用自动制备色谱仪(洗脱剂:1~5%乙酸乙酯/石油醚)纯化,收集合并纯净组分后浓缩蒸干得到(苯并噻吩-5-基)-氨基甲酸叔丁酯(18)(5.58g,22.38mmol,57%收率,灰白色固体)。
[0210]1h nmr(400mhz,cdcl3):δ=8.00(s,1h),7.75(d,j=8.8hz,1h),7.43(d,j=5.6hz,1h),7.26(d,j=5.6hz,1h),7.20(dd,j=8.4,2.0hz,1h),6.58(brs,1h),1.54(s,9h).
[0211]
步骤12.2化合物19的制备
[0212]
在-60℃氮气氛围下向(苯并噻吩-5-基)-氨基甲酸叔丁酯(18)(3.00g,12.03mmol)的无水四氢呋喃(55ml)溶液中持续滴加正丁基锂(14.4ml,2.5m己烷溶液)达30分钟。反应液维持在-60℃下继续搅拌60分钟后持续滴加硼酸三异丙酯(4.6ml,d=0.818g/ml)达30分钟,随后自然升至室温并持续搅拌18小时。所得黄色浊液用0.4m稀盐酸(90ml)淬灭后用乙酸乙酯(60ml
×
3)萃取。合并有机相后用饱和食盐水(100ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干得到5-(叔丁氧羰酰)氨基苯并噻吩-2-硼酸(19)(4.07g,约80%纯度,92%收率,灰白色固体)并直接用于下一步反应。
[0213]1h nmr(400mhz,dmso-d6):δ=9.42(brs,1h),8.44(d,j=1.6hz,1h),8.05(brs,1h),7.87-7.79(m,2h),7.42-7.35(m,1h),7.34(s,1h),1.49(s,9h).
[0214]
步骤12.3化合物20的制备
[0215]
向n-(5-溴-[1,2,4]三唑并[1,5-a]吡啶-2-基)-环丙甲酰胺(int-1)(967mg,3.44mmol)和5-(叔丁氧羰酰)氨基苯并噻吩-2-硼酸(19)(2.16g,约80%纯度,5.88mmol)的1,4-二氧六环(27.5ml)和水(5.5ml)混合溶液中在氮气流下加入1,1
’-
双(二苯基膦)二茂铁二氯化钯(151mg,0.206mmol)和磷酸钾(1.97g,9.29mmol)。该混合液用氮气置换数次后,在氮气氛围下加热至95℃并持续搅拌20小时。所得深棕色悬浊液(含小试批次eb3-19)冷却
后加水(55ml)稀释。过滤收集所生成的沉淀并用水(10ml
×
2)洗涤,再真空干燥得到{5-[5-(叔丁氧羰酰)氨基苯并噻吩-2-基]-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(20)(2.38g,约70%纯度粗品,棕色固体)并直接用于下一步反应。
[0216]
lcms:tr=0.975,1.111min in 5-95ab_1.5min_220&254.lcm,ms(esi)m/z=450.1[m+h]
+
.
[0217]1h nmr(400mhz,dmso-d6):δ=11.21(brs,1h),8.76(s,1h),8.04(d,j=8.8hz,1h),7.86(s,1h),7.81-7.68(m,3h),7.45-7.37(m,1h),3.27(s,3h),2.17-2.04(m,1h),1.41(s,9h),1.40-1.29(m,4h).
[0218]
步骤12.4化合物exp-28的制备
[0219]
在10℃下将{5-[5-(叔丁氧羰酰)氨基苯并噻吩-2-基]-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(20)(2.38g,约70%纯度粗品)悬浮于无水二氯甲烷(20ml)中并向其中滴加三氟乙酸(10ml,d=1.54g/ml)。所得溶液在10℃下持续搅拌2小时后,用氮气流鼓泡法除去大部分挥发物,再用1m碳酸钾溶液(50ml)处理。过滤收集所生成的沉淀并用二氯甲烷/甲醇(10ml,体积比1:1)打浆处理。所得固体经真空干燥得到{5-(5-氨基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(exp-28)(1.60g粗品,约70%纯度,93%的两步收率,黄色固体)并未经纯化直接用于下一步反应。
[0220]
lcms:tr=0.780min in 5-95ab_1.5min_220&254_shimadzu.lcm,ms(esi)m/z=350.0[m+h]
+
.
[0221]1h nmr(400mhz,dmso-d6):δ=11.19(brs,1h),8.63(s,1h),7.80-7.62(m,4h),7.60-7.45(m,2h),5.24(brs,2h),2.23-1.97(m,1h),1.38-1.17(m,4h).
[0222]
步骤12.5化合物exp-29的制备
[0223]
在15℃下向{5-(5-氨基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(exp-28)(100mg粗品,约70%纯度)和三乙胺(0.1ml,d=0.727g/ml)的无水二氯甲烷(4ml)浊液中加入乙酸酐(30μl,d=1.09g/ml)。反应液在15℃下持续搅拌20小时,将所得黄棕色悬浊液减压浓缩除去大部分挥发物后加水(8ml)稀释。过滤收集生成的沉淀并用水(2ml
×
2)洗涤,再用高效液相色谱(30-60%乙腈/0.04%氨水+10mm碳酸氢铵)制备分离,收集合并纯净组分,减压浓缩并冻干后得到最终产物{5-(5-乙酰胺基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(exp-29)(11.2mg,100%纯度,14%收率,白色固体)。
[0224]
lcms:tr=0.877min in 5-95 ab_1.5min_220&254_shimadzu.lcm,ms(esi)m/z=392.1[m+h]
+
.
[0225]
tr=3.715min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=392.2[m+h]
+
.
[0226]
hplc:tr=2.52min in 10-80 cd_8min.met(xbridge shield rp18 5um).
[0227]1h nmr(400mhz,dmso-d6):δ=11.24(brs,1h),10.15(brs,1h),8.83(s,1h),8.41(s,1h),7.99(d,j=7.2hz,1h),7.84-7.66(m,3h),7.52(d,j=7.2hz,1h),2.26-2.16(m,1h),2.10(s,3h),1.01-0.72(m,4h).
[0228]
步骤12.6化合物exp-14的制备
[0229]
在0℃下向{5-(5-氨基苯并噻吩-2-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙
甲酰胺(exp-28)(100mg粗品,约70%纯度)的无水n,n-二甲基甲酰胺(1.8ml)溶液中加入钠氢(27mg,60wt%于矿物油中)。0℃下搅拌30分钟后滴加2-甲氧基乙烷磺酰氯(sm22)(57mg,0.349mmol)。反应液升至15℃并持续搅拌14小时后,用1m氯化铵水溶液(10ml)淬灭,而后用乙酸乙酯(12ml,10ml和8ml)萃取。合并有机相,依次用水(10ml
×
2)和饱和食盐水(15ml)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。粗品用薄层制备板(展开剂:二氯甲烷/乙酸乙酯=1/3)分离得到橙色油状物,随后再用高效液相色谱(35-65%乙腈/0.04%氨水+10mm碳酸氢铵)制备分离,收集合并纯净组分,减压浓缩并冻干后得到最终产物{5-[5-(2-甲氧基)乙烷磺酰胺基苯并噻吩-2-基]-[1,2,4]三唑并[1,5-a]吡啶-2-基}环丙甲酰胺(exp-30)(2.9mg,98.09%纯度,3%收率,灰白色固体)。
[0230]
lcms:tr=1.340min in 5-95 ab_2min_220&254_shimadzu.lcm,ms(esi)m/z=472.2[m+h]
+
.
[0231]
tr=5.648min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=472.1[m+h]
+
.
[0232]
hplc:tr=3.33min in 10-80 ab_8min.met.
[0233]1h nmr(400mhz,dmso-d6):δ=11.24(brs,1h),8.80(s,1h),8.02(d,j=8.8hz,1h),7.86-7.65(m,4h),7.31(dd,j=8.4,2.0hz,1h),3.68(t,j=6.0hz,2h),3.46(t,j=6.0hz,2h),3.19(s,3h),2.23-2.01(m,1h),0.98-0.78(m,4h).
[0234]
步骤12.7化合物exp-31的制备
[0235]
cis-n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基)-2-氟-环丙甲酰胺(int-2)(97mg,0.32mmol)和[5-(甲烷磺酰胺)苯并噻吩-2-基]硼酸(20)(110mg,0.41mmol)溶于二氧六环(2ml)和水(0.4ml)中,在氮气氛围中快速加入碳酸钾(168mg,1.22mmol)和1,1-双(二苯基磷)二茂铁氯化钯(30mg,0.041mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用hplc纯化(分离柱:ageladurashell c18 250*25mm*10um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 35-65%)纯化,并得到白色目标产物2-氟-n-[5-[5-(甲烷磺酰胺)苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-31)(12.9mg,收率7%,纯度96.66%)。
[0236]
lcms:tr=3.732min in 10-80ab_7min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=446.2[m+h]
+
.
[0237]
hplc:tr=1.81min in 10-80cd_8min.met,chromatography(xbridge shield rp18 5um).
[0238]
1h nmr(400mhz,dmso-d6):δ=11.31(s,1h),9.46(s,1h),8.81(s,1h),8.07(d,j=8.8hz,1h),7.86-7.60(m,4h),7.35(d,j=8.8hz,1h),5.16-4.76(m,1h),3.03(s,3h),1.82-1.63(m,1h),1.32-1.15(m,2h).
[0239]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表5的实施例化合物。
[0240]
【表5】
[0241][0242]
实施例13:化合物exp-34的制备
[0243]
合成路线如下:
[0244][0245]
步骤13.1化合物21的制备
[0246]
5-溴苯并噻吩(sm9)(200mg,0.94mmol)和吗啡啉(sm23)(163mg,1.88mmol)溶解于n,n-二甲基乙酰胺(4ml)中,向其中加入碘化亚铜(18mg,0.094mmol)和叔丁醇钠(180mg,1.88mmol)且在190℃下微波反应2小时。反应液和eb5-63(100mg 5-溴苯并噻吩)合并处理,用乙酸乙酯(20ml)和水(20ml)稀释。水相用乙酸乙酯(20ml
×
3)萃取,有机相用饱和食盐水(20ml
×
3)洗涤,无水硫酸钠干燥后过滤,浓缩后用硅胶柱层析(洗脱剂:0%~20%乙酸乙酯/石油醚)得到淡黄色目标产物4-(苯并噻吩-5-基)吗啡啉(21)(80mg,收率24%,纯度92%)。
[0247]
lcms:tr=0.975min in 5-95ab_1.5min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=220.0[m+h]
+
.
[0248]
步骤13.2化合物22的制备
[0249]
4-(苯并噻吩-5-基)吗啡啉(21)(80mg,0.36mmol)溶解在四氢呋喃(2ml)中,在-75
℃氮气氛围下向其中缓慢滴加正丁基锂(2.5m in hexane,0.44ml)。搅拌60分钟后,加入硼酸三异丙酯(137mg,0.73mmol),然后缓慢升到15-20℃并持续搅拌16小时。反应液用10%盐酸溶液(10ml)淬灭且水相用乙酸乙酯(10ml
×
3)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩得到黄色固体(5-吗啡啉苯并噻吩-2-基)硼酸(22)(60mg,粗品)。
[0250]
步骤13.3化合物exp-34的制备
[0251]
n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(int-1)(30mg,0.11mmol)和(5-吗啡啉苯并噻吩-2-基)硼酸(22)(42mg,0.16mmol)溶于二氧六环(0.5ml)和水(0.1ml)中,在氮气氛围中快速加入碳酸钾(44mg,0.32mmol)和1,1-双(二苯基磷)二茂铁氯化钯(8mg,0.01mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用乙酸乙酯(20ml)和水(20ml)稀释,水相用乙酸乙酯(20ml
×
3)萃取,有机相用饱和食盐水(20ml
×
3)洗涤,无水硫酸钠干燥,过滤,浓缩后用制备hplc纯化(分离柱:ageladurashell c18 150*25mm*5um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 40-70%)纯化得到白色固体n-[5-[5-吗啡啉苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-34)(1.9mg,收率4%,纯度91.88%)。
[0252]
lcms:tr=2.719min in 10-80ab_4min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=420.3[m+h]
+
.
[0253]
hplc:tr=3.07min(10-80ab_8min.met),chromatography.
[0254]
1h nmr(400mhz,dmso-d6):δ=11.20(s,1h),8.71(s,1h),7.91(d,j=8.8hz,1h),7.78-7.69(m,3h),7.37(d,j=1.6hz,1h),7.28(dd,j=8.8and 2.0hz,1h),3.79(t,j=4.8hz,4h),3.19(t,j=4.8hz,4h),2.18-2.02(m,1h),0.92-0.86(m,4h).
[0255]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表6的实施例化合物。
[0256]
【表6】
[0257]
[0258][0259]
实施例14:化合物exp-37的制备
[0260][0261]
步骤14.1化合物23的制备
[0262]
在氩气流下向盛有5-溴苯并噻吩(sm9)(3.74g,17.20mmol)和三乙胺(5.0ml,d=0.727g/ml,35.92mmol)的无水乙醇(99ml)和四氢呋喃溶液(33ml)的钢化玻璃瓶中加入1,1
’-
双(二苯基膦)二茂铁二氯化钯(730mg,1.00mmol)。钢化玻璃瓶加盖气压表头后,用氩气置换内压数次,随后充入45psi内压的一氧化碳。反应在80℃和45psi内压的一氧化碳气氛下搅拌50小时。所得深红色溶液冷却后减压蒸去大部分乙醇,余下的n,n-二甲基甲酰胺溶液加叔丁基甲醚稀释(300ml)后,用水(100ml
×
3)和饱和食盐水(120ml)洗涤,经无水硫酸钠干燥,过滤后减压浓缩。残留物用自动制备色谱仪(洗脱剂:1~3.5%乙酸乙酯/石油醚)纯化,收集合并纯净组分后浓缩蒸干得到苯并噻吩-5-甲酸乙酯(23)(3.27g,92%收率,淡黄色油状物)。
[0263]1h nmr(400mhz,cdcl3):δ=8.54(d,j=1.2hz,1h),8.01(dd,j=8.4,1.6hz,1h),7.92(d,j=8.4hz,1h),7.51(d,j=5.6hz,1h),7.43(d,j=5.6hz,1h),4.42(q,j=7.2hz,2h),1.43(t,j=7.2hz,3h).
[0264]
步骤14.2化合物24的制备
[0265]
向苯并噻吩-5-甲酸乙酯(23)(2.83g,13.72mmol)的四氢呋喃(16ml)溶液中加入一水合氢氧化锂(1.17g,27.9mmol)的水(14ml)溶液,所得混合物升温至55℃并持续搅拌8小时。所得无色乳浊液冷却后减压浓缩以除去大部分挥发物,在搅拌下用1m盐酸(29ml)酸化后,用乙酸乙酯(60ml
×
2)萃取。合并萃取相并用饱和食盐水(60ml)洗涤,经无水硫酸钠干燥,过滤并减压浓缩得到苯并噻吩-5-甲酸(24)(2.39g,98%收率,灰白色固体)并直接用
于下一步反应。
[0266]1h nmr(400mhz,dmso-d6):δ=12.96(brs,1h),8.51(s,1h),8.12(d,j=8.4hz,1h),7.90(dd,j=8.0,1.2hz,1h),7.88(d,j=6.0hz,1h),7.61(d,j=5.6hz,1h).
[0267]
步骤14.3化合物25的制备
[0268]
在-60℃氮气氛围下向苯并噻吩-5-甲酸(24)(597mg,3.35mmol)的无水四氢呋喃(15ml)溶液中持续滴加叔丁基锂(7.8ml,1.3m己烷溶液)达10分钟。移除低温浴,任反应自然升温至0℃并持续搅拌40分钟后,所得黄色悬浊液反应液重新降温至-60℃并向其中持续滴加硼酸三异丙酯(1.2ml,d=0.818g/ml)达10分钟,随后任凭反应液自然升至室温(15℃)并持续搅拌15小时。所得黄色溶液以0.2m稀盐酸(60ml)淬灭后用乙酸乙酯(20ml,15ml和10ml)萃取。合并萃取相用饱和食盐水(25ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干得到浅红色固体。该固体用正己烷/氯仿/甲醇混合体系(18ml,体积比30/5/1)打浆,过滤收集不溶物,以正己烷(3ml
×
2)洗涤后真空干燥得到2-双羟硼基苯并噻吩-5-甲酸(25)(505mg,约90%纯度,61%收率,灰白色固体)并直接用于下一步反应。
[0269]1h nmr(400mhz,dmso-d6):δ=12.98(brs,1h),8.59(brs,2h),8.52-8.46(m,1h),8.08(d,j=8.4hz,1h),8.06(s,1h),7.93-7.87(m,1h).
[0270]
步骤14.4化合物exp-37的制备
[0271]
在氮气流下向n-(5-溴-[1,2,4]三唑并[1,5-a]吡啶-2-基)-环丙甲酰胺(int-1)(369mg,1.31mmol)和2-双羟硼基苯并噻吩-5-甲酸(25)(505mg,约90%纯度,2.05mmol)的1,4-二氧六环(10ml)和水(2ml)混合溶液中加入磷酸钾(822mg,3.87mmol)和1,1
’-
双(二苯基膦)二茂铁二氯化钯(32mg,0.043umol)。该混合液用氮气置换数次后,在氮气氛围下加热至95℃并持续搅拌20小时。所得深棕色悬浊液(含小试批次eb3-34)冷却后加1m盐酸酸化至ph=3后以乙酸乙酯(25ml,20ml和15ml)萃取。合并萃取相用饱和食盐水(30ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干。粗品先用自动制备色谱仪(洗脱剂:80~99%乙酸乙酯/石油醚,再1~8%甲醇/二氯甲烷)纯化。收集所需馏分并减压蒸干得到粗品的浅黄色固体(155mg)。
[0272]
lcms:tr=0.946min in 5-95 ab_1.6min_220&254_shimadzu.lcm,ms(esi)m/z=401.0[m+na]
+
.
[0273]
tr=3.973min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=379.2[m+h]
+
.
[0274]
上述固体中的73毫克用高效液相色谱(25-55%乙腈/0.225%甲酸)制备分离,收集合并纯净组分,减压浓缩并冻干后得到最终产物2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]苯并噻吩-5-甲酸(exp-37)(6.5mg,93%纯度,2%收率,灰白色固体)。
[0275]
lcms:tr=3.787min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=379.2[m+h]
+
.
[0276]
hplc:tr=3.79min in 0-60 ab_8min.met.
[0277]1h nmr(400mhz,dmso-d6):δ=11.22(brs,1h),8.89(s,1h),8.55(s,1h),8.21(d,j=8.4hz,1h),8.00(d,j=8.4hz,1h),7.88-7.70(m,3h),2.18-1.93(m,1h),0.91-0.84(m,4h).
[0278]
实施例15:化合物exp-38的制备
[0279]
合成路线如下:
[0280][0281]
步骤15.1化合物26的制备
[0282]
在氩气流下向盛有6-溴苯并噻吩(sm6)(935mg,4.30mmol)和三乙胺(1.3ml,d=0.727g/ml,9.34mmol)的无水乙醇(25ml)和四氢呋喃溶液(10ml)的钢化玻璃瓶中加入1,1
’-
双(二苯基膦)二茂铁二氯化钯(170mg,0.232mmol)。钢化玻璃瓶加盖气压表头后,用氩气置换内压数次,随后充入50psi内压的一氧化碳。反应在80℃和50psi内压的一氧化碳气氛下搅拌40小时。所得深红色溶液冷却后减压蒸去大部分乙醇,余下的n,n-二甲基甲酰胺溶液加叔丁基甲醚稀释(90ml)后,用水(30ml
×
3)和饱和食盐水(40ml)洗涤,经无水硫酸钠干燥,过滤后减压浓缩。残留物用自动制备色谱仪(洗脱剂:1~3%乙酸乙酯/石油醚)纯化,收集合并纯净组分后浓缩蒸干得到苯并噻吩-6-甲酸甲酯(26)(821mg,99%收率,白色固体)。
[0283]1h nmr(400mhz,cdcl3):δ=8.62(s,1h),8.03(d,j=8.0hz,1h),7.86(d,j=8.4hz,1h),7.65(d,j=5.6hz,1h),7.40(d,j=5.6hz,1h),3.96(s,3h).
[0284]
步骤15.2化合物27的制备
[0285]
将苯并噻吩-6-甲酸甲酯(26)(821mg,4.27mmol),频那醇硼烷(sm10)(0.75ml,d=0.882g/ml,4.91mmol),甲氧基(环辛二烯)合铱(i)二聚体(45mg,0.064mmol)和4,4
’-
二叔丁基-2,2
’-
联吡啶(39mg,0.141mmol)的正己烷混合液(24ml)置换氮气氛围数次并在15℃下持续搅拌12小时。所得红色溶液过滤除去不溶物后减压浓缩。残留物用自动制备色谱仪(洗脱剂:1~4%乙酸乙酯/石油醚)纯化,收集合并纯净组分后浓缩蒸干得到6-甲氧羰基苯并噻吩-2-硼酸频那醇酯(27)(450mg,33%收率,灰白色固体)。
[0286]1h nmr(400mhz,dmso-d6):δ=8.63(s,1h),8.00(dd,j=8.4,1.6hz,1h),7.91(s,1h),7.88(d,j=8.4hz,1h),3.96(s,3h),1.38(s,12h).
[0287]
步骤15.3化合物28的制备
[0288]
向28毫升微波管中依次加入n-(5-溴-[1,2,4]三唑并[1,5-a]吡啶-2-基)-环丙甲酰胺(int-1)(183mg,0.651mmol)和6-甲氧羰基苯并噻吩-2-硼酸频那醇酯(27)(290mg,0.911mmol)的1,4-二氧六环(5.5ml)和水(1ml)混合溶液中加入碳酸钠(138mg,1.30mmol),随即在氮气流下加入双(三苯基膦)二氯化钯(59mg,0.085mmol)。向该微波管内用鼓泡法置换氮气氛围后,密封置于微波反应仪上,在110℃下持续搅拌反应2.5小时。所得橙色悬浊液冷却后减压浓缩除去大部分挥发物。残留物加二氯甲烷/甲醇溶解(50ml,体积比4/1)并过
滤除去不溶物。滤液减压浓缩蒸干后,所得粗品(含小试批次eb3-71)用自动制备色谱仪(洗脱剂:0~25%丙酮/(石油醚/乙酸乙酯=1/1体积比))纯化。收集所需馏分并减压蒸干得到2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]苯并噻吩-6-甲酸甲酯(28)(90mg,60%纯度,19%收率,粉色固体)。
[0289]
lcms:tr=1.073min in 5-95 ab_1.6min_220&254_shimadzu.lcm,ms(esi)m/z=393.1[m+h]
+
.
[0290]
tr=3.580min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=393.2[m+h]
+
.
[0291]
步骤15.4化合物29的制备
[0292]
向2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]苯并噻吩-6-甲酸甲酯(28)(90mg,约60%纯度,0.138mmol)的四氢呋喃(1.4ml)溶液中加入一水合氢氧化锂(29mg,0.691mmol)的水(0.6ml)溶液。反应液在30℃下持续搅拌12小时。所得棕色溶液浓缩除去大部分挥发物后用1m盐酸溶液酸化至ph=3。过滤收集产生的沉淀并用水(0.5ml
×
2)洗涤,然后真空干燥得到2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]苯并噻吩-6-甲酸(29)(66mg,约50%纯度,63%收率,粉色固体)并直接用于下一步反应。
[0293]
lcms:tr=1.028min in 5-95 ab_2min_220&254_shimadzu.lcm,ms(esi)m/z=379.2[m+h]
+
.
[0294]
步骤15.5化合物exp-38的制备
[0295]
向2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]苯并噻吩-6-甲酸(29)(66mg,约50%纯度,0.087mmol)的无水n,n-二甲基甲酰胺(1.4ml)溶液中依次加入o-(7-氮杂苯并三氮唑-1-基)-n,n,n’,n
’-
四甲基脲六氟磷酸盐(50mg,0.132mmol)和n,n-二异丙基乙胺(0.05ml,d=0.742g/ml,0.287mmol),稍后加入(2-噻唑-2-基)乙胺(17mg,0.133mmol)。反应液在15℃下搅拌6小时后,所得黄色浆状物用乙酸乙酯(15ml)稀释后,以水(5ml
×
2)和饱和食盐水(5ml)洗涤,经无水硫酸钠干燥,过滤并浓缩蒸干。残留物用高效液相色谱(25-55%乙腈/0.04%氨水)制备分离,收集纯净组分,减压浓缩并冻干后得到最终产物2-[2-(环丙甲酰胺基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]-n-(2-噻唑-2-基)-乙基]苯并噻吩-6-甲酰胺(exp-38)(4.8mg,100%纯度,11%收率,白色固体)。
[0296]
lcms:tr=2.820min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=489.3[m+h]
+
.
[0297]
tr=2.860min in 10-80 ab_7min_220&254_shimadzu.lcm,ms(esi)m/z=489.3[m+h]
+
.
[0298]
hplc:tr=2.55min in 10-80 ab_8min.met.
[0299]1h nmr(400mhz,methanol-d4):δ=8.86(s,1h),8.40(s,1h),8.01(d,j=8.4hz,1h),7.84(d,j=8.4,1.2hz,1h),7.78-7.69(m,3h),7.67(dd,j=7.6and 2.0hz,1h),7.49(d,j=3.2hz,1h),3.82(t,j=6.8hz,2h),3.41(t,j=6.8hz,2h),2.13-1.92(m,1h),1.15-1.05(m,2h),1.03-0.93(m,2h).
[0300]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表7的实施例化合物。
[0301]
【表7】
[0302][0303][0304]
实施例16:化合物exp-41的制备
[0305]
合成路线如下:
[0306][0307]
步骤16.1化合物30的制备
[0308]
[5-(甲磺胺基)苯并噻吩-2-基]硼酸(3)(1.1g,4.1mmol)、5-溴-[1,2,4]三唑[1,5-a]吡啶-2-胺(sm12)(432mg,2.0mmol)和碳酸钠(430mg,4.1mmol)溶于水(5ml)和四氢呋喃(20ml)氮气置换3次后加入1,1-双(二苯基磷)二茂铁氯化钯(285mg,0.41mmol)再氮气置换3次,100℃下反应1小时。加入乙酸乙酯(10ml)和水(10ml)稀释,然后乙酸乙酯(10ml
×
3)萃取,无水硫酸钠干燥,过滤,真空浓缩,柱层析纯化,得到黄色固体n-(2-(2-氨基-[1,2-4]三唑并[1,5-a]吡啶-5-基)苯并噻吩-5-基]甲磺酰胺(30)(700毫克,粗品)。
[0309]
步骤16.2化合物31的制备
[0310]
n-[2-(2-氨基-[1,2,4]三唑[1,5-a]吡啶-5-基)苯并噻吩-5-基]甲磺酰胺(30)(200mg,0.56mmol)和亚硝酸钠(77mg,1.1mmol)溶于乙腈(2ml)中。0℃氮气保护下搅拌0.1小时,然后滴加溴化氢(287mg,1.7mmol),20℃搅拌0.9小时。加入水(5ml),饱和碳酸钾调节至ph=7,二氯甲烷(10ml
×
3)萃取。饱和食盐水(10ml)洗涤,无水硫酸钠干燥,过滤,浓缩,得到黄色固体化合物n-[2-(2-溴-[1,2,4]三唑[1,5-a]吡啶-5-基)苯并噻吩-5-基]甲磺酰胺(31)(150mg,粗品)。
[0311]
步骤16.3化合物exp-41的制备
[0312]
将n-[2-(2-溴-[1,2,4]三唑[1,5-a]吡啶-5-基)苯并噻吩-5-基]甲磺酰胺(31)(20mg,0.05mmol)、1-甲基吡唑-3-胺(9mg,0.09mmol)、brettphos pd g3(9mg,0.01mmol)和叔丁醇钠(36mg,0.38mmol)溶于1,4-二氧六环(2ml)。微波150℃下加热0.2小时。乙酸乙酯(10ml)和水(10ml)萃取。有机层无水硫酸钠干燥,过滤,浓缩,粗品制备分离得到黄色固体n-[2-[2-[(1-甲基吡唑-3-基)氨基]-[1,2,4]三唑[1,5-a]吡啶-5-基]苯并噻吩-5-基]甲磺酰胺(exp-41)(6.7mg,30%产率)。
[0313]
lcms:tr=2.133min in 10-80ab_4min_220&254_agilent.met,chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=440.3[m+h]
+
.
[0314]
hplc:tr=2.16min in 10-80cd_8min.met,chromatography(xbridge c18 2.1*50mm 5um).
[0315]
1h nmr(400mhz,dmso-d6):δ=11.25(s,1h),9.87(s,1h),8.81(s,1h),8.05(d,j=8.8hz,1h),7.86-7.65(m,4h),7.34(d,j=8.8hz,1h),6.83(s,1h),3.84(s,3h),3.10(s,3h).
[0316]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表8的实施例化合物。
[0317]
【表8】
[0318][0319][0320]
实施例17:化合物exp-44的制备
[0321]
合成路线如下:
[0322][0323]
步骤17.1化合物32的制备
[0324]
5-溴苯并噻吩(sm9)(4g,18.77mmol),乙烯基三氟硼酸钾盐(5.03g,37.54mmol),1,1-双(二苯基磷)二茂铁氯化钯(686mg,0.938mmol)和碳酸铯(15.29g,46.93mmol)溶于水(4ml)/二氧六环(40ml)中并置换氮气三次后再90度下搅拌2小时。该混合物加水(50ml)并且用二氯甲烷(50ml
×
3)萃取。有机相用饱和食盐水(80ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:1%乙酸乙酯在石油醚中)得到无色油状5-乙烯基苯并噻吩(32)(2.97g,收率98%)。
[0325]
1h nmr(400mhz,dmso-d6):δ=7.87-7.80(m,2h),7.49(dd,j=8.4,2.0hz,1h),7.45(d,j=5.6hz,1h),7.34(d,j=5.2hz,1h),6.92-6.80(m,1h),5.83(d,j=17.2hz,1h),5.30(d,j=11.2hz,1h).
[0326]
步骤17.2化合物33的制备
[0327]
5-乙烯基苯并噻吩(32)(2.97g,18.54mmol)溶于四氢呋喃(60ml)中加入9-硼双环[3.3.1]壬烷(0.5m溶于四氢呋喃,55.6ml)在0度下,该反应体系在10-15度下搅拌个16小时后再加水(6ml)、3m氢氧化钠溶液(30ml)和过氧化氢(30ml,30wt%),该反应体系在50度下搅拌个2小时。该混合物用饱和的亚硫酸钠(40ml)淬灭后通过乙酸乙酯(60ml
×
3)萃取,有机相用饱和食盐水(80ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:50%乙酸乙酯在石油醚中)得到白色固体2-(苯并噻吩-5-基)乙醇(33)(2.4g,收率72%)。
[0328]
1h nmr(400mhz,dmso-d6):δ=7.84(d,j=8.4hz,1h),7.70(d,j=0.8hz,1h),7.45(d,j=5.6hz,1h),7.31(d,j=5.2hz,1h),7.24(dd,j=8.4,1.6hz,1h),3.93(t,j=6.0hz,2h),3.00(t,j=6.4hz,2h),1.48-1.40(m,1h).
[0329]
步骤17.3化合物34的制备
[0330]
2-(苯并噻吩-5-基)乙醇(33)(1g,5.61mmol)溶于四氢呋喃(10ml)中在0度下加入钠氢(448mg,11.22mmol)后并且在15度下搅拌半小时,4-甲基苯磺酰氯(1.28g,6.73mmol)加入到反应体系中并且在10-15度下搅拌1.5小时。该混合物用水(25ml)淬灭后通过乙酸乙酯(40ml
×
2)萃取,有机相用饱和食盐水(40ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:5%乙酸乙酯/石油醚)得到白色固体2-(苯并噻吩-5-基)乙基4-甲基苯磺酸(34)(1.4g,收率75%)。
[0331]
1h nmr(400mhz,dmso-d6):δ=7.74(d,j=8.0hz,1h),7.61(d,j=8.4hz,2h),
7.54(d,j=0.8hz,1h),7.45(d,j=5.6hz,1h),7.24(d,j=5.6hz,1h),7.16(d,j=8.0hz,2h),7.08(dd,j=8.4,1.6hz,1h),4.27(t,j=6.8hz,2h),3.07(t,j=6.8hz,2h),2.39(s,3h).
[0332]
步骤17.4化合物35的制备
[0333]
2-(苯并噻吩-5-基)乙基4-甲基苯磺酸(34)(400mg,1.20mmol)溶于n,n-二甲基甲酰胺(5ml)中加入碳酸钾(498mg,3.61mmol)和硫代吗啡啉-1,1-二氧化物(406mg,3.01mmol)并且在85度下搅拌2小时。该混合物加入乙酸乙酯(20ml)后用饱和食盐水(20ml)洗3次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:50%乙酸乙酯/石油醚)得到白色固体4-[2-(苯并噻吩-5-基)乙基]-硫代吗啡啉-1,1-二氧化物(35)(80mg,收率19%)。
[0334]
lcms:tr=0.898min in 10-80ab_2.0min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=296.1[m+h]
+
.
[0335]
步骤17.5化合物36的制备
[0336]
4-[2-(苯并噻吩-5-基)乙基]-硫代吗啡啉-1,1-二氧化物(35)(70mg,0.236mmol)溶于四氢呋喃(2ml)中在氮气环境下降温到-75度后缓慢加入正丁基锂(2.5m溶于正己烷,0.28ml)并且在-75度下搅拌半小时,加入硼酸三异丙酯(133mg,0.71mmol)后升到15-20度并且搅拌2小时。该混合物用1m盐酸(10ml)淬灭后通过乙酸乙酯(20ml
×
3)萃取,有机相用饱和食盐水(20ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩得到白色固体[5-[2-(1,1-二氧化物-1,4-硫代吗啡啉-4-基)乙基]苯并噻吩-2-基]苯硼酸(36)(70mg,收率87%)。
[0337]
lcms:tr=0.788min in 10-80ab_2.0min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=340.1[m+h]
+
.
[0338]
步骤17.6化合物exp-44的制备
[0339]
n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(int-1)(58mg,0.206mmol),[5-[2-(1,1-二氧化物-1,4-硫代吗啡啉-4-基)乙基]苯并噻吩-2-基]苯硼酸(36)(70mg,0.206mmol),磷酸钾(109mg,0.515mmol)和1,1-双(二苯基磷)二茂铁氯化钯(7mg,0.01mmol)溶于二氧六环(2ml)/水(0.2ml)中并置换氮气三次后再90度下搅拌2小时。该反应混合物加水(30ml)并且用二氯甲烷(30ml
×
3)萃取,有机相用饱和食盐水(50ml)洗一次然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:5%甲醇/二氯甲烷)得到粗品后用碱性制备分离纯化得到白色固体目标产物n-[5-[2-(1,1-二氧化物-1,4-硫代吗啡啉-4-基)乙基]苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(exp-44)(9.2mg,收率9%)。
[0340]
lcms:tr=2.360min in 10-80ab_7min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=496.4[m+h]
+
.
[0341]
hplc:tr=2.76min in 10-80cd_8min.met,chromatography(xbridge shield rp18 5um).
[0342]
1h nmr(400mhz,dmso-d6):δ=11.21(s,1h),8.80(s,1h),7.99(d,j=8.0hz,1h),7.84(s,1h),7.80-7.67(m,3h),7.39(d,j=7.6hz,1h),3.13-3.06(m,4h),3.05-2.96(m,4h),2.95-2.85(m,2h),2.84-2.75(m,2h),2.18-2.03(m,1h),0.96-0.82(m,4h).
[0343]
实施例18:化合物exp-41的制备
[0344][0345]
步骤18.1化合物37的制备
[0346]
2-(苯并噻吩-5-基)乙基4-甲基苯磺酸(34)(320mg,0.962mmol)溶于乙腈(5ml)中加入碳酸钾(399mg,2.89mmol)和4,4-二氟哌啶(sm8)(233mg,1.93mmol)并且在85度下搅拌16小时。该混合物加入水(20ml)后用乙酸乙酯(20ml
×
3)萃取,有机相用饱和食盐水(20ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:10%乙酸乙酯/石油醚)得到白色固体1-[2-(苯并噻吩-5-基)乙基]-4,4-二氟哌啶(37)(300mg,收率82%)。
[0347]
lcms:tr=0.985min in 5-95ab_1.5min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*30mm),ms(esi)m/z=282.0[m+h]
+
.
[0348]
步骤18.2化合物38的制备
[0349]
1-[2-(苯并噻吩-5-基)乙基]-4,4-二氟哌啶(37)(65mg,0.231mmol)溶于四氢呋喃(2ml)中在氮气环境下降温到-75度后缓慢加入正丁基锂(2.5m溶于正己烷,0.277ml)并且在-75度下搅拌半小时,加入硼酸三异丙酯(130mg,0.693mmol)后升到15-20度并且搅拌2小时。该混合物用1m盐酸(10ml)淬灭后通过乙酸乙酯(20ml
×
3)萃取,有机相用饱和食盐水(20ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩得到白色固体[5-[2-(4,4-二氟哌啶)乙基]苯并噻吩-2-基]苯硼酸(38)(70mg,收率93%)。
[0350]
lcms:tr=0.197min in 5-95ab_4.0min_220&254_shimadzu.lcm chromatography(xtimate,2.1*30mm,3um b:xbrige shield 2.1*50mm),ms(esi)m/z=326.1[m+h]
+
.
[0351]
步骤18.3化合物exp-45的制备
[0352]
n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(38)(60mg,0.215mmol),[5-[2-(4,4-二氟哌啶)乙基]苯并噻吩-2-基]苯硼酸(70mg,0.215mmol),磷酸钾(114mg,0.538mmol)和1,1-双(二苯基磷)二茂铁氯化钯(7mg,0.01mmol)溶于二氧六环(2ml)/水(0.2ml)中并置换氮气三次后再90度下搅拌2小时。该反应混合物加水(30ml)并且用二氯甲烷(30ml
×
3)萃取,有机相用饱和食盐水(50ml)洗一次然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:5%甲醇/二氯甲烷)得到粗品后用碱性制备分离纯化得到白色固体目标产物n-[5-[5-[2-(4,4-二氟哌啶)乙基]苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(exp-45)(8.5mg,收率8%)。
[0353]
lcms:tr=2.494min in 10-80ab_7min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=482.4[m+h]
+
.
[0354]
hplc:tr=3.41min in 10-80cd_8min.met,chromatography(xbridge shield rp 18 5um).
[0355]
1h nmr(400mhz,dmso-d6):δ=11.21(s,1h),8.79(s,1h),7.98(d,j=8.4hz,1h),
7.83(s,1h),7.79-7.68(m,3h),7.37(dd,j=8.0,0.8hz,1h),2.96-2.85(m,2h),2.70-2.65(m,2h),2.63-2.58(m,4h),2.16-2.05(m,1h),2.0-1.85(m,4h),0.94-0.84(m,4h).
[0356]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备方法而制备以下表9的实施例化合物。
[0357]
【表9】
[0358][0359][0360]
实施例19:化合物exp-48的制备
[0361]
合成路线如下:
[0362][0363]
步骤19.1化合物39的制备
[0364]
将5-溴苯并噻吩(sm9)(1.5g,7.04mmol)溶于二甲基甲酰胺(13.5ml)和吡啶(0.75ml),加入氰化亚铜(851mg,9.50mmol)。反应液在氮气氛围下于150度搅拌20小时。反应液倒入含有n,n-二甲基乙二胺(1ml)的水(20ml)溶液中。水相用乙酸乙酯(80ml
×
3)中,有机相合并,食盐水(20ml
×
3)洗涤,用无水硫酸钠干燥,过滤,真空浓缩,粗品通过硅胶柱(乙酸乙酯/石油醚,从0%到5%)纯化得到5-氰基苯并噻吩(39)(579mg,51%收率,99%纯度,黄色固体)。
[0365]
1h nmr(400mhz,dmso-d6):δ=8.43(d,j=0.8hz,1h),8.25(d,j=8.0hz,1h),7.99(d,j=5.6hz,1h),7.72(dd,j=8.4,1.6hz,1h),7.58(d,j=5.6hz,1h).
[0366]
步骤19.2化合物40的制备
[0367]
将四氢铝锂(552mg,14.55mmol)溶于四氢呋喃(20ml)中,零度下加入5-氰基苯并噻吩(579mg,3.64mmol)的四氢呋喃(10ml)溶液,反应液于室温下搅拌16小时。反应液零度下加入水(0.6ml)和氢氧化钠溶液(15wt%,0.6ml)和水(1.8ml)淬灭,搅拌0.5小时,加入无水硫酸钠干燥,加入乙酸乙酯(100ml)。反应液过滤,有机相食盐水(20ml)洗涤,无水硫酸钠干燥,过滤,真空浓缩,得到苯并噻吩-5-基甲胺(40)(590mg,90%收率,91%纯度,黄色固体),直接用于下一步。
[0368]
1h nmr(400mhz,dmso-d6):δ=7.90(d,j=8.4hz,1h),7.81(d,j=0.4hz,1h),7.72(d,j=5.2hz,1h),7.41(d,j=5.2hz,1h),7.34(dd,j=8.4,1.2hz,1h),3.82(s,2h).
[0369]
步骤19.3化合物41的制备
[0370]
将苯并噻吩-5-基甲胺(40)(590mg,3.61mmol)溶于二氯甲烷(10ml)中,零度下加入三乙胺(1.28g,12.64mmol)和甲磺酸酐(1.26g,7.22mmol),反应液于室温下搅拌16小时。反应液零度下加入饱和碳酸氢钠(20ml)淬灭,用二氯甲烷(50ml
×
3)萃取。有机相合并,用食盐水(50ml)洗涤,无水硫酸钠干燥,过滤,真空浓缩,粗品经过硅胶色谱柱(乙酸乙酯/石油醚,从0%到39%)纯化得到n-(苯并噻吩-5-基甲基)甲磺酰胺(41)(655mg,75%收率,淡黄色固体)。
[0371]
1h nmr(400mhz,dmso-d6):δ=7.98(d,j=8.4hz,1h),7.84(s,1h),7.77(d,j=5.2hz,1h),7.61(t,j=6.4hz,1h),7.46(d,j=5.6hz,1h),7.35(dd,j=8.4,1.6hz,1h),4.27(d,j=6.4hz,2h),2.86(s,3h).
[0372]
步骤19.4化合物42的制备
[0373]
将n-(苯并噻吩-5-基甲基)甲磺酰胺(41)(200mg,0.829mmol)溶于四氢呋喃(4ml)中,-78℃下,氮气保护下,加入正丁基锂(2.5m in hexane,1.33ml),搅拌0.5小时,然后加入硼酸三异丙酯(655mg,3.48mmol),反应液于-78℃下搅拌0.5小时,升至室温,搅拌2小时。反应液用稀盐酸(1m,5ml)零度下淬灭,室温搅拌半个小时。反应液用乙酸乙酯(40ml
×
3)萃取。有机相合并,用食盐水(30ml)洗涤,无水硫酸钠干燥,过滤,真空浓缩,得到[5-(甲磺酰胺甲基)苯并噻吩-2-基]硼酸(42)(260mg,粗品,黄色固体),直接用于下一步。
[0374]
步骤19.5化合物exp-48的制备
[0375]
将n-(5-溴-[1,2,4]三氮唑[1,5-a]吡啶-2-基)环丙基甲酰胺(int-)(100mg,0.356mmol),碳酸钾(98mg,0.711mmol)和[5-(甲磺酰胺甲基)苯并噻吩-2-基]硼酸(42)(142mg,0.498mmol)溶于二氧六环(2ml)和水(0.4ml)中,氮气置换三次,然后加入1,1-双(二苯基磷)二茂铁氯化钯(39mg,0.0534mmol),再次氮气置换三次,反应液于95度搅拌2小时。反应液浓缩得到粗品经过硅胶色谱柱纯化(甲醇/二氯甲烷,从0%到3%)得到n-[5-[5-(甲磺酰胺甲基)苯并噻吩-2-基]-[1,2,4]三氮唑[1,5-a]吡啶-2-基]环丙基甲酰胺(exp-48)(30.2mg,19%收率,98.30%纯度,黄色固体)。
[0376]
lcms:tr=2.890min in 10-80ab_7min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*3mm,3um),ms(esi)m/z=442.3[m+h]
+
.
[0377]
hplc:tr=3.09min in 10-80ab_8min_215&220&254.met.
[0378]
1h nmr(400mhz,dmso-d6):δ=11.23(brs,1h),8.86(s,1h),8.07(d,j=8.4hz,1h),7.94(s,1h),7.79-7.71(m,3h),7.68(t,j=6.4hz,1h),7.47(dd,j=8.4,1.2hz,1h),4.32(d,j=6.4hz,2h),2.90(s,3h),2.20-2.02(m,1h),0.95-0.82(m,4h).实施例20:化合物exp-49的制备
[0379]
合成路线如下:
[0380][0381]
步骤20.1化合物43的制备
[0382]
苯并噻吩-5-胺(1)(200mg,1.34mmol),三乙胺(406mg,4.02mmol)和4-二甲基氨基吡啶(16mg,0.13mmol)溶解在二氯甲烷(5ml)中,在0℃下加入环丙基磺酰氯(sm16)(282mg,2.01mmol)后在15-25℃下搅拌2小时。反应液浓缩旋干采用硅胶柱层析(洗脱剂:0%~50%乙酸乙酯/石油醚)得到白色目标产物n-(苯并噻吩-5-基)环丙磺酰胺(43)(200mg,收率59%)。
[0383]
1h nmr(400mhz,dmso-d6):δ=9.74(s,1h),7.94(d,j=8.4hz,1h),7.81-7.69(m,2h),7.44(d,j=5.6hz,1h),7.31-7.23(m,1h),2.65-2.55(m,1h),0.97-0.82(m,4h).
[0384]
步骤20.2化合物44的制备
[0385]
n-(苯并噻吩-5-基)环丙磺酰胺(43)(200mg,0.79mmol)溶解在四氢呋喃(3ml)中,在-75℃氮气氛围下向其中缓慢滴加正丁基锂(2.5m in hexane,1.26ml)。搅拌60分钟后,
加入硼酸三异丙酯(297mg,1.58mmol),然后缓慢升到15-20℃并持续搅拌2小时。反应液用10%的盐酸溶液(20ml)淬灭且水相用乙酸乙酯(20ml
×
3)萃取,有机相用无水硫酸钠干燥,过滤,滤液旋干得到淡黄色固体[5-(环丙磺酰胺)苯并噻吩-2-基]硼酸(44)(200mg,粗品)。
[0386]
步骤20.3化合物exp-49的制备
[0387]
n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(int-1)(80mg,0.28mmol)和[5-(环丙磺酰胺)苯并噻吩-2-基]硼酸(44)(169mg,0.57mmol)溶于二氧六环(3ml)和水(0.6ml)中,在氮气氛围中快速加入碳酸钾(118mg,0.85mmol)和1,1-双(二苯基磷)二茂铁氯化钯(20mg,0.028mmol)。混合物经氮气置换三次,然后在90℃下搅拌2小时。反应混合物过滤,滤液用二氯甲烷(20ml)和水(20ml)稀释,水相用二氯甲烷(20ml
×
3)萃取,有机相用无水硫酸钠干燥后过滤浓缩旋干后采用硅胶柱层析(洗脱剂:0%~10%甲醇/二氯甲烷)和hplc纯化(分离柱:waters xbridge 150*25mm*5um;流动相:a:水(0.04%nh3.h2o+10mm nh4hco3)和b:乙腈;梯度:b 30-60%)纯化得到白色固体n-[5-[5-(环丙磺酰胺)苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基]环丙甲酰胺(exp-49)(10.4mg,收率7%,纯度84.89%)。
[0388]
lcms:tr=2.188min in 10-80ab_4min_220&254_shimadzu.lcm(xtimate c18,3um,2.1*3mm),ms(esi)m/z=454.30[m+h]
+
.
[0389]
hplc:tr=3.35min in 10-80ab_8min.met,chromatography.
[0390]
1h nmr(400mhz,dmso-d6):δ=11.22(s,1h),9.82(s,1h),8.81(s,1h),8.05(d,j=8.8hz,1h),7.83(d,j=2.0hz,1h),7.79-7.70(m,3h),7.38(dd,j=8.8,2.0hz,1h),2.74-2.61(m,1h),2.18-2.01(m,1h),0.97-0.83(m,8h).
[0391]
实施例21:化合物exp-50的制备
[0392]
合成路线如下:
[0393][0394]
步骤21.1化合物45的制备
[0395]
2-[5-(溴甲基)苯并噻吩-嚬哪醇硼酸酯(14)(200mg,0.566mmol)溶于乙腈(3ml)中加入碳酸钾(156mg,1.13mmol)和1-(2-羟基乙基)哌嗪(1a)(147mg,1.13mmol)并且在35搅拌2小时。该混合物加入水(20ml)后用乙酸乙酯(30ml
×
3)萃取,有机相用饱和食盐水(30ml)洗一次,然后用无水硫酸钠干燥后过滤浓缩得到红色固体2-[4-[[2-(嚬哪醇硼酸酯-2-基)苯并噻吩-5-基]甲基哌嗪-1-基]乙醇(45)(80mg,收率35%)。
[0396]
lcms:tr=0.837min in 5-95ab_2.0min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*30mm),ms(esi)m/z=403.2[m+h]
+
.
[0397]
步骤21.2化合物exp-50的制备
[0398]
n-(5-溴-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(45)(50mg,0.177mmol),2-[4-[[2-(嚬哪醇硼酸酯-2-基)苯并噻吩-5-基]甲基哌嗪-1-基]乙醇(71mg,0.177mmol),磷
酸钾(75mg,0.355mmol)和1,1-双(二苯基磷)二茂铁氯化钯(6mg,0.008mmol)溶于二氧六环(1ml)/水(0.1ml)中并置换氮气三次后再70度下搅拌2小时。该反应混合物加水(30ml)并且用二氯甲烷(30ml
×
3)萃取,有机相用饱和食盐水(50ml)洗一次然后用无水硫酸钠干燥后过滤浓缩。得到的粗品用硅胶柱层析(洗脱剂:10%甲醇/二氯甲烷)得到粗品后用碱性制备分离纯化得到白色固体目标产物n-[5-[5-[[4-(2-羟乙基)哌嗪-1基)甲基]苯并噻吩-2-基]-[1,2,4]三唑[1,5-a]吡啶-2-基)环丙甲酰胺(exp-50)(2.0mg,收率2.0%)。
[0399]
lcms:tr=1.539min in 10-80ab_4min_220&254_shimadzu.lcm chromatography(xtimate c18,3um,2.1*3mm),ms(esi)m/z=477.3[m+h]
+
.
[0400]
hplc:tr=2.40min in 10-80cd_8min.met,chromatography(xbridge shield rp18 5um).
[0401]
1h nmr(400mhz,dmso-d6):δ=11.20(s,1h),8.81(s,1h),8.02(d,j=8.0hz,1h),7.86(s,1h),7.80-7.68(m,3h),7.42(dd,j=8.4,0.8hz,1h),4.39(s,1h),3.61(s,2h),3.49-3.47(m,2h),2.48-2.34(m,10h),2.18-2.05(m,1h),0.93-0.84(m,4h).
[0402]
实施例22:化合物exp-51的制备
[0403]
合成路线如下:
[0404][0405]
步骤22.1化合物51的制备
[0406]
将6-溴吡啶-2-胺(sm13)(5g,28.90mmol)和溴代丙酮酸乙酯(sm14)(6.20g,31.79mmol)溶于乙醇(30ml),于85度下搅拌16小时。反应液过滤,滤饼收集,用石油醚/乙酸乙酯(5/1,v/v,50ml)打浆,固体过滤,干燥得到5-溴咪唑[1,2-a]吡啶-2-羧酸甲酯(51)(8.41g,83%yield,溴化氢盐,黄色固体)。
[0407]1h nmr(400mhz,dmso-d6):δ=8.60-8.50(m,1h),7.82-7.76(m,1h),7.65-7.47(m,2h),4.38(q,j=7.2hz,2h),1.35(t,j=7.2hz,3h).
[0408]
步骤22.2化合物52的制备
[0409]
将5-溴咪唑[1,2-a]吡啶-2-羧酸甲酯(51)(2g,5.71mmol,溴化氢盐)溶于水,四氢呋喃和甲醇各5毫升溶剂中,加入一水合氢氧化锂(839mg,20.00mmol)。反应液于15度搅拌2小时。反应液加水(10ml),用二氯甲烷(20ml)。水相用稀盐酸(1n)调至ph=5,过滤,滤饼干燥得到5-溴咪唑[1,2-a]吡啶-2-甲酸(52)(1.24g,90%收率,白色固体)用于下一步。
[0410]
lcms:tr=0.342min in 5-95ab_1.5min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*30mm,3um),ms(esi)m/z=242.9[m+2+h]
+
.
[0411]
步骤22.3化合物53的制备
[0412]
将5-溴咪唑[1,2-a]吡啶-2-甲酸(27)(1.24g,5.14mmol),4a分子筛(2.48g,10.29mmol)和三乙胺(1.56g,15.43mmol)悬浮于叔丁醇(12ml)中,加入叠氮磷酸二苯酯(2.12g,7.72mmol)。反应液于氮气氛围下在90度搅拌16小时。反应液过滤,滤液真空浓缩,粗品通过硅胶色谱纯化(甲醇/二氯甲烷,从0%到2%)得到5-溴咪唑[1,2-a]吡啶-2-胺基甲酸叔丁酯(53)(1.01g,49%收率,78%纯度,黄色固体)。
[0413]
lcms:tr=1.443min in 10-80ab_2min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*3mm,3um),ms(esi)m/z=312.1[m+h]
+
.
[0414]
步骤22.4化合物54的制备
[0415]
将5-溴咪唑[1,2-a]吡啶-2-胺基甲酸叔丁酯(53)(1.01g,3.24mmol)溶于二氯甲烷(20ml)中加入三氟乙酸(12.32g,108.05mmol),反应液于15度搅拌2小时。反应液浓缩,用饱和碳酸氢钠调节ph=9,然后用二氯甲烷(30ml
×
3)萃取。有机相合并,用食盐水(20ml)洗涤,无水硫酸钠干燥,过滤,浓缩得到粗品,通过硅胶色谱柱纯化(甲醇/二氯甲烷,从0%到5%)得到5-溴咪唑[1,2-a]吡啶-2-胺(54)(400mg,58%收率,黄色固体)。
[0416]
1h nmr(400mhz,dmso-d6):δ=7.24(d,j=8.0hz,1h),7.06(d,j=7.2hz,1h),7.02-6.97(m,2h),5.30(brs,2h).
[0417]
步骤22.5化合物46的制备
[0418]
将5-溴咪唑[1,2-a]吡啶-2-胺(54)(400mg,1.89mmol)溶于n,n-二甲基乙酰胺(2ml)中加入环丙基甲酰氯(237mg,2.26mmol),于15度下搅拌2小时。反应液加入饱和碳酸氢钠(20ml)淬灭,过滤,滤饼水洗(10ml),干燥得到n-(5-溴咪唑[1,2-a]吡啶-2-基)环丙基甲酰胺(46)(460mg,87%收率,黄色固体)。
[0419]
lcms:tr=1.263min in 5-95ab_2min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*3mm,3um),ms(esi)m/z=280.0[m+h]
+
.
[0420]
步骤22.6化合物exp-51的制备
[0421]
将[5-(甲磺酰胺基)苯并噻吩-2-基]硼酸(3)(242mg,0.892mmol),n-(5-溴咪唑[1,2-a]吡啶-2-基)环丙基甲酰胺(46)(100mg,0.357mmol)和碳酸钾悬浮于二氧六环(2ml)和水(0.4ml)中,用氮气置换三次,然后加入1,1-双(二苯基磷)二茂铁氯化钯(39mg,0.0535mmol),用氮气置换三次,100度搅拌2小时。反应液真空浓缩得到粗品,用硅胶色谱柱纯化(甲醇/二氯甲烷,从0%到4%)得到粗品,进一步用制备hplc(column:ageladurashell c18 150*25mm*5um;condition:water(0.04%nh
3.
h2o+10mm nh4hco3)-acn;begin:b 30%,end b 60%;gradient time:(8min);flowrate(25ml/min))纯化得到n-[5-[5-(甲磺酰胺基)苯并噻吩-2-基]咪唑[1,2-a]吡啶-2-基]环丙基甲酰胺(exp-51)(10.2mg,6%收率,94.5%纯度,黄色固体)。
[0422]
lcms:tr=3.255min in 10-80ab_7min_220&254_shimadzu.lcm,chromatography(xtimate c18 2.1*3mm,3um),ms(esi)m/z=427.2[m+h]
+
.
[0423]
hplc:tr=2.75min in 10-80ab_8min.met.
[0424]
1h nmr(400mhz,dmso-d6):δ=11.12(s,1h),9.90(br s,1h),8.37(s,1h),8.09-8.01(m,2h),7.84(d,j=2.0hz,1h),7.55(d,j=8.8hz,1h),7.41-7.31(m,2h),7.18(d,j=7.2hz,1h),3.02(s,3h),1.98-1.88(m,1h),0.85-0.74(m,4h).
[0425]
依据与上述实施例相同的方法,使用市售化合物或参考所示的中间化合物的制备
方法而制备以下表10的实施例化合物。
[0426]
【表10】
[0427][0428]
实施例23:jak激酶抑制活性检测
[0429]
将上述实施例的化合物应用于对jak激酶抑制活性的检测和筛选。
[0430]
1.方法
[0431]
在第一块96孔板上每孔加入相应体积dmso。每孔分别加入20~30μl的100~200μm不同供试化合物储存液。震荡混合3min。对所有化合物进行3x系列稀释。在第二块96孔板上每孔加入60~80μl kinase buffer,从第一块96孔板每孔取1~2μl溶液加入在第二块96孔板相应孔中。震荡混合3min。从第二块化合物稀释板每孔取5μl化合物稀释液转移到384孔测试板相应孔。在测试板每孔加1~2μl tk substrate
–
biotin。每孔加1~2μl酶混合液。同时设置不加酶的blank孔。每孔加1~2μl atp溶液,封好板子后室温进行反应。反应时间分别为:jak1:2小时,jak2:30min,jak3:30min,tyk2:50min。每孔加3~5μlof streptavidin-xl665,3~5μl of tk antibody-cryptate,封好板子后室温放置30分钟以结束反应。perkinelmer envision机器上读取665nm和620nm的fluorescence。
[0432]
ic50计算
[0433]
计算每个孔的htrf ratio:(665signal/620signal)x 10^4.
[0434]
抑制百分率基于以下公式计算:
[0435]
抑制%=(ratio
max-ratio
化合物
)/(ratio
max-ratio
min
)]
×
100%
[0436]
其中ratio
化合物
为给定化合物浓度下的htrf ratio,ratio
min
为加入blank孔的htrfratio,ratio
max
为不加入化合物的情况下的htrf ratio。通过使用graphpad prism 5.0软件,使用非线性回归模型绘制s型剂量-抑制率曲线并计算ic
50
值。
[0437]
filgotinib表示具有如下结构的化合物:
[0438][0439]
2.实验结果
[0440]
各送检化合物测得的ic
50
(nm)检测值如下表11所示。
[0441]
【表11】化合物对jak的抑制活性
[0442]
[0443]
[0444][0445]
备注:ic
50
值在0-10nm标记为a;10-30nm标记为b;30-50nm标记为c;50-500nm标记为d;大于500nm标记为e;nt代表为未测试。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1