一种新的依托咪酯杂质及制备方法和其应用与流程
1.本发明属于药物合成领域,具体涉及一种新的依托咪酯杂质及制备方法和其应用。
背景技术:
2.依托咪酯(etomidate)是用于全身麻醉诱导的静脉麻醉药,根据文献报道(现代应用药学,1997,(1):30-31,19970201;us3354173a,19671121),依托咪酯产业化制备路线如下:
[0003][0004]
上述合成路线中,所述步骤5,即依托咪酯的合成反应中,一旦步骤1的产物pm-1存在残留,而残留的pm-1在步骤5中参与反应就会产生亚硝胺类化合物。亚硝胺类化合物具有致癌风险,属于人用药物注册技术要求国际协调会议(ich)《评估和控制药物中dna反应性(致突变)杂质以限制潜在致癌风险》指南中提及的“关注队列”物质。
[0005]
因此,非常有必要对其产生的亚硝胺化合物杂质(如本发明式ⅰ所示结构的杂质)进行监控,以保障依托咪酯制剂安全性。
技术实现要素:
[0006]
本发明的目的在于提供一种如式ⅰ所示结构的新的依托咪酯杂质,
[0007][0008]
本发明的目的之一还在于提供式ⅰ杂质的制备方法,包括下述步骤:称取所需量的(r)-2-((1-苯乙基)氨基)乙酸乙酯与亚硝基化试剂,在酸性试剂存在下发生反应,生成式ⅰ杂质,
[0009][0010]
本发明的优选技术方案中,所述反应在有机溶剂体系下进行,所述有机溶剂选自乙腈、二氯甲烷、dmf、氯仿的任一种或其组合。
[0011]
本发明的优选技术方案中,所述亚硝基化试剂选自亚硝酸钠、亚硝酸钾、亚硝酸乙酯、亚硝酸甲酯、亚硝酸的任一种或其组合。
[0012]
本发明的优选技术方案中,反应物料(r)-2-((1-苯乙基)氨基)乙酸乙酯:亚硝基化试剂摩尔比为1:2-1:6,优选为1:3-1:5。
[0013]
本发明的优选技术方案中,所述酸性试剂选自有机酸或无机酸。
[0014]
本发明的优选技术方案中,所述有机酸选自对甲苯磺酸、苯甲酸、三氟乙酸、三氯乙酸的任一种或其组合;所述无机酸选自盐酸、硫酸、硝酸的任一种或其组合。
[0015]
本发明的优选技术方案中,反应物料(r)-2-((1-苯乙基)氨基)乙酸乙酯:酸性试剂摩尔比为1:1-1:3,优选为1:2。
[0016]
本发明的优选技术方案中,所述反应温度为10℃-50℃,优选为20℃-30℃。
[0017]
本发明的优选技术方案中,所述反应时间不低于5小时,优选不低于8小时。
[0018]
本发明的优选技术方案中,式ⅰ杂质经分离后,制得。
[0019]
本发明的优选技术方案中,所述分离步骤为萃取、浓缩。
[0020]
本发明的优选技术方案中,所述萃取溶剂选自乙酸乙酯、氯仿、二氯甲烷、乙酸异丙酯的任一种或其组合。
[0021]
本发明的优选技术方案中,所述浓缩方式选自减压浓缩或常压浓缩。
[0022]
本发明的优选技术方案中,式ⅰ杂质经分离、纯化后,制得。
[0023]
本发明的优选技术方案中,所述纯化步骤为柱层析或减压蒸馏。
[0024]
本发明的优选技术方案中,所述柱层析步骤包括:(1)将式ⅰ杂质粗品加入硅胶,溶于溶剂1中,浓缩至固体;(2)将步骤(1)所述固体加入至硅胶柱中,依次用溶剂2、溶剂3洗脱;(3)收集洗脱液,减压浓缩,即得。
[0025]
本发明的优选技术方案中,所述硅胶选自100-400目,优选为300-400目。
[0026]
本发明的优选技术方案中,所述柱层析的溶剂1选自乙酸乙酯、二氯甲烷、乙腈、甲醇、丙酮的任一种或其组合。
[0027]
本发明的优选技术方案中,所述柱层析的溶剂2选自正己烷、正庚烷、石油醚的任一种或其组合。
[0028]
本发明的优选技术方案中,所述柱层析的溶剂3选自乙酸乙酯、正己烷、石油醚、丙酮、正庚烷的任一种或其组合。优选为乙酸乙酯:正己烷、乙酸乙酯:正庚烷、丙酮:正己烷、丙酮:石油醚体积比为1:5-1:15的混合物,优选为1:10。
[0029]
本发明的目的之一还在于提供一种式ⅰ杂质用作标准品或对照品中的应用。
[0030]
本发明的优选技术方案中,所述式ⅰ用作标准品或对照品用于定性或定量检测依托咪酯或其中间体的质量和纯度。
[0031]
本发明的优选技术方案中,所述定量检测方法采用高效液相色谱-质谱法。
[0032]
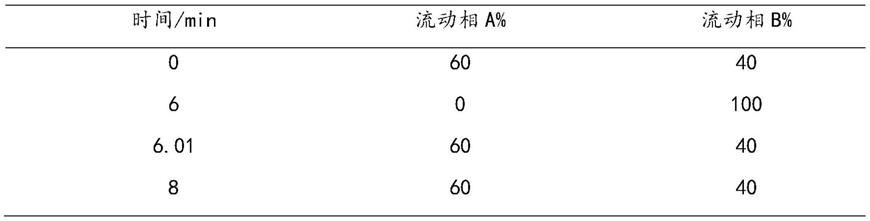
本发明的优选技术方案中,所述高效液相色谱-质谱法测定条件如下:用十八烷基硅烷键合硅胶为填充剂;以0.1%甲酸水为流动相a,以甲醇为流动相b,进行如下表所示的线性梯度洗脱,流速为每分钟0.1-1ml;柱温为20-40℃;选择质荷比237.0/105.0作为检测离子对。
[0033][0034]
本发明的优选技术方案中,所述流速为每分钟0.5ml;柱温为30℃。
[0035]
本发明的目的还在于提供一种安全性高的依托咪酯药物组合物,所述药物组合物中含有依托咪酯或其药学上可接受的盐和含量不高于1ppm的式ⅰ杂质。
[0036]
本发明的优选技术方案中,所述药物组合物中式ⅰ杂质含量不高于0.4ppm。
[0037]
本发明的优选技术方案中,所述药学上可接受的盐选自甲磺酸盐、乙磺酸盐、苯磺酸盐、富马酸盐、酒石酸盐、氢溴酸盐、磷酸盐、盐酸盐、硫酸盐的任一种。
[0038]
本发明的目的之一还在于提供一种安全性高的依托咪酯药物制剂,所述制剂由高安全的依托咪酯药物组合物与药学上可接受的载体组成,其中,所述药物组合物中含有依托咪酯或其药学上可接受的盐和含量不高于1ppm的式ⅰ杂质。
[0039]
本发明的优选技术方案中,所述制剂组合物中的式ⅰ杂质含量不高于0.4ppm。
[0040]
本发明的优选技术方案中,所述的药物制剂为注射剂,更优选为注射乳剂。
[0041]
本发明的目的之一还在于提供一种具有麻醉活性的药物包,所述的药物包包含所述安全性高的依托咪酯药物组合物或制剂及其他药物。
[0042]
本发明的优选技术方案中,所述的其他药物选自利多卡因、布托啡诺、丙泊酚、东良菪碱、咪达唑仑、舒芬太尼、瑞芬太尼、米索前列醇、咪唑安定、潘库溴胺、阿托品、氯胺酮、硫喷妥钠的任一种或其组合。
[0043]
除非另有说明,本发明涉及液体与液体之间的百分比时,所述的百分比为体积/体积百分比;本发明涉及液体与固体之间的百分比时,所述百分比为体积/重量百分比;本发明涉及固体与液体之间的百分比时,所述百分比为重量/体积百分比;其余为重量/重量百分比。
[0044]
与现有技术相比,本发明具有下述有益技术效果:
[0045]
本发明首次制备并表征且确认具有致癌风险的如式ⅰ所示结构的亚硝胺类杂质,将式ⅰ杂质用作对照品或标准品用于控制依托咪酯及其制备中间体的纯度和质量,进而保障依托咪酯的纯度和质量,以保障药品质量,从而保障临床用药安全和患者生命健康。
附图说明
[0046]
图1式ⅰ杂质的1h nmr图谱
[0047]
图2式ⅰ杂质的
13
c nmr图谱
[0048]
图3式ⅰ杂质的ms图谱
[0049]
图4式ⅰ杂质的对照品高效液相色谱-质谱图谱
[0050]
图5式ⅰ杂质用作对照品检测依托咪酯有关物质的高效液相色谱-质谱图谱(样品1)
具体实施方式
[0051]
下面列举一部分具体实施例对本发明进行说明,有必要在此指出的是以下具体实施例只用于对本发明作进一步说明,不代表对本发明保护范围的限制。其他人根据本发明做出的一些非本质的修改和调整仍属于本发明的保护范围。
[0052]
实施例1式ⅰ杂质的制备
[0053]
于20-30℃,将5g(r)-2-((1-苯乙基)氨基)乙酸乙酯中加入50ml乙腈溶解,加入6.66g亚硝酸钠、9.18g对甲苯磺酸,保持温度20-30℃搅拌14h,tlc检测反应完毕。在反应液中加入纯化水(100g),用乙酸乙酯(60ml*2)萃取,合并有机相,有机相用纯化水(100ml)洗涤,收集有机相。有机相于45-50℃减压浓缩,得到黄色液体。
[0054]
将浓缩的黄色液体采用干法上样,加入硅胶(300-400目)10g,加入50ml乙酸乙酯,于45-50℃减压浓缩。柱层析柱中加入硅胶(300-400目)50g。先用正己烷(200ml)洗脱,再用乙酸乙酯:正己烷=1:10(550ml)洗脱,收集洗脱液,洗脱液于45-50℃减压浓缩,得到2.09g黄色液体,收率36.7%,hplc纯度(方法如下)96.5%。
[0055]
纯度测定:取上述反应产物适量,精密称定,加乙醇-水(50:50)溶解并定量稀释成每1ml中约含2mg的溶液,作为供试品溶液。照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(ymc c
18
100mm
×
4.6mm,3μm),以5g/l碳酸铵溶液为流动相a,乙腈为流动相b,按照下表1进行梯度洗脱,流速为每分钟2.0ml,柱温为35℃,检测波长为210nm。精密量取供试品溶液10μl,注入液相色谱仪,记录色谱图,供试品溶液色谱图中,按面积归一化法计算,主峰纯度不得低于95.0%。
[0056]
表1
[0057][0058]
结构鉴定见图1-3:
[0059]1h nmr(600mhz,dmso)δ1.100~1.124ppm(3h,t,j=7.2hz),δ1.808~1.820ppm(3h,d,j=7.2hz),δ3.982~4.019ppm(2h,m),δ4.055~4.082ppm(1h,d,j=16.2h),δ4.187~4.214ppm(1h,d,j=16.2h),δ5.839~5.875ppm(1h,m),δ7.329~7.349(1h,m),δ7.353~7.713(4h,m)。
[0060]
13
c nmr(600mhz,dmso):δ13.84ppm,δ18.85ppm,δ45.41ppm,δ60.75ppm,δ61.75ppm,δ127.17ppm(2c),δ128.09ppm,δ128.66ppm(2c),δ139.18ppm,δ165.45pp。
[0061]
ms:[m+h]
+
=236.9。
[0062]
实施例2式ⅰ杂质用作对照品检测依托咪酯有关物质
[0063]
取不同批次(3批)的依托咪酯样品适量,精密称定,加甲醇溶液溶解并定量稀释制成每1ml中约含100mg的溶液,作为供试品溶液;另取实施例合成的式ⅰ杂质适量,精密称定,加甲醇溶液溶解并定量稀释制成每1ml中约含40ng的溶液,作为对照品溶液。照高效液相色谱-质谱法测定,用十八烷基硅烷键合硅胶为填充剂(3.0mm
×
100mm,3.5μm或性能相当的色谱柱);以0.1%甲酸水为流动相a,以甲醇为流动相b,按下表2进行线性梯度洗脱,流速为每分钟0.5ml;柱温为30℃。采用质谱检测器,电喷雾正离子模式(esi
+
),进行多反应监测(mrm),选择质荷比237.0/105.0作为检测离子对(源温度为550℃;离子喷雾电压为5500伏)。精密量取供试品溶液及对照品溶液5μl,分别注入液相色谱-质谱联用仪,记录色谱图,供试品溶液色谱图中,如有式1杂质峰,按外标法以峰面积计算,不得过0.4ppm。
[0064]
表2
[0065][0066]
式ⅰ杂质测定结果见表3:
[0067]
表3式ⅰ杂质用作对照品检测依托咪酯有关物质的结果
[0068]
项目质荷比样品1样品2样品3式1杂质237.0/105.0未检出未检出未检出
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1