一种具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点及其制备方法和应用与流程
本发明属于纳米材料技术领域,涉及一种具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点及其制备方法和应用。具体而言,通过微波加热鱼精蛋白与乙二醇的混合溶液,获得大量具有细胞核仁靶向能力的碳量子点,并可以应用于活细胞中的细胞核仁成像和核酸递送转染。
背景技术:
细胞核仁是真核细胞的重要细胞器,与dna复制、mrna和核糖体合成以及基因表达密切相关,它的状态被视为实时监测细胞行为和恶性转化的细胞指标。然而,动态追踪活细胞中的细胞核并不容易,因为这种成像探针必须满足以下要求:细胞毒性小;能够跨越包括细胞膜与核膜在内的种种细胞屏障;能够特异地集聚在核仁内;光稳定性好,能够经受连续光或激光的长期照射。因此,制备一种符合这些要求的新型荧光探针显得极为迫切。
近年来,荧光碳点因为具有合成快速、生物相容性高、抗光漂白和发射光谱可调谐等独特性能,被广泛应用于生物成像领域。然而,被细胞内化后,除非经过特殊修饰,它们主要定位在细胞质而非细胞核中。核定位信号(nls)是一种由精氨酸或赖氨酸组成的核靶向序列,常常作为配体修饰纳米颗粒,辅助入核。鱼精蛋白也属于nls材料,受到fda批准,曾被用于运载质粒入核。但是,至今无人采用这种便宜、安全、天然的材料修饰碳量子点。
技术实现要素:
为了克服现有技术的缺点与不足,本发明的首要目的在于提供一种具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点的制备方法。
本发明的另一目的在于提供通过上述制备方法制备得到的具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点。该荧光碳量子点具有便宜、安全、合成快速的特点。
本发明的另一目的在于提供上述具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点的应用。
本发明的另一目的在于提供一种基于该荧光碳量子点的活细胞核成像的方法。
本发明的另一目的在于提供一种基于该荧光碳量子点的核酸递送转染的方法。
本发明的目的通过下述技术方案实现:
一种具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点的制备方法,包括如下步骤:
通过微波加热鱼精蛋白盐与乙二醇的混合溶液,透析提纯,获得具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点。
优选的,所述的鱼精蛋白盐选自硫酸鱼精蛋白和盐酸鱼精蛋白;
优选的,所述的微波加热的条件为700w加热7~8min;进一步为700w加热7.5min;
优选的,所述的鱼精蛋白盐与乙二醇的比值为0.8~1g:100ml;进一步为1g:100ml;
优选的,所述的透析所用的透析膜为8000~10000mwco透析膜;进一步为10000mwco透析膜;
优选的,所述的透析的时间为2~5天;进一步为2天。
具体包括如下步骤:
将乙二醇和溶于超纯水的鱼精蛋白盐溶液混合,涡旋以形成均匀系统;微波加热;冷却至室温后,产物全部加入透析膜中,用超纯水透析,获得澄清淡黄色水溶液,即具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点溶液,置于室温保存。
优选的,所述的溶于超纯水的鱼精蛋白盐溶液中鱼精蛋白盐与超纯水的比值为0.8~1.5g:60ml;进一步为1g:60ml;
优选的,所述的涡旋的时间为10s~60s;进一步为10s。
优选的,所述的微波加热是置于家用微波炉中加热;微波功率可根据所需的光致发光强度进行调整,优选为700w;所述的加热时间亦之,优选为7.5min。
优选的,所述的超纯水为18.2mω·cm的超纯水;
一种具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点,通过上述制备方法制备得到。
所述的具有核仁靶向能力的鱼精蛋白修饰的荧光碳量子点在活细胞中细胞核仁成像或核酸递送转染中的应用。
一种基于鱼精蛋白修饰的荧光碳量子点的活细胞核仁成像的方法,包括以下步骤:
将细胞接种于玻璃皿中,密度为12500个细胞/孔,培养24小时,然后加入鱼精蛋白修饰的荧光碳量子点浓度为10~35μg/ml(优选为30μg/ml)的无血清dmem培养基进行染色,然后置于共焦激光扫描显微镜的活细胞培养箱中,继续培养,采集荧光图像。
优选的,所述的细胞为hek293细胞,但不限于此。
优选的,所述的继续培养的时间为3~12小时;进一步为3小时。
其中,图像成像时间可根据需要进行调整,在每30分钟扫描一次的情况下,至少可以维持12小时。
一种基于鱼精蛋白修饰的荧光碳量子点的核酸递送转染的方法,包括以下步骤:
将细胞接种于24孔微孔板,密度为8000个细胞/孔,培养24小时,吸出培养基,加入新鲜无血清dmem培养基和含有dna与荧光碳量子点的复合物溶液;孵育4小时,吸出培养基,用pbs洗涤两次,加入新鲜dmem培养基。培养24小时,用倒置荧光显微镜观察并拍摄,且用流式细胞仪检测转染细胞数占总细胞数的比例。
优选的,所述的细胞为hek293细胞,但不限于此。
优选的,所述的dna为编码绿色荧光蛋白的质粒;进一步的,所述的编码绿色荧光蛋白的质粒为pegfp-n1质粒;
优选的,所述的含有dna与荧光碳量子点的复合物溶液中荧光碳量子点的浓度为60μg/ml,dna与荧光碳量子点的重量比为1~1.5:3;进一步优选为1.5:3;
优选的,所述的新鲜无血清dmem培养基和含有dna和荧光碳量子点的复合物溶液的体积比为9:1;具体分别取450μl和50μl。
本发明相对于现有技术具有如下的优点及效果:
(1)选择鱼精蛋白作为表面钝化配体,通过微波法一步制备具有核仁靶向能力的荧光碳量子点,便宜、安全、快速;制备工艺简单、容易操作且成本低。
(2)该荧光碳量子点作为细胞探针具有光稳定性强、生物相容性高、多色荧光等优点,可以穿透细胞屏障并特异性靶向细胞核仁,且实现了对活细胞核仁的长时间实时成像。
(3)本荧光碳量子点可用于建立荧光碳量子点/质粒纳米复合物,进行体外转染,在基因递送方面具有潜在的应用价值;说明该荧光碳量子点有潜力成为一种具有核仁靶向能力的多功能荧光探针。
附图说明
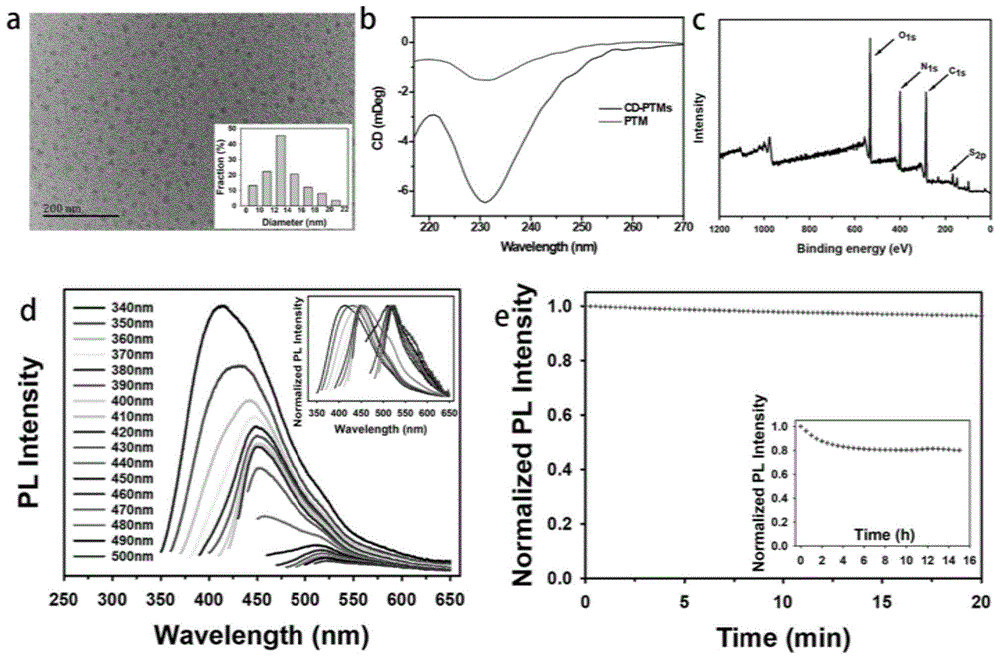
图1是实施例1的附图,是鱼精蛋白修饰的荧光碳量子点的表征及光学性质;其中,a为该荧光碳量子点的tem图像和粒径分布情况;b为该荧光碳量子点(cd-ptms)和硫酸鱼精蛋白(ptm)水溶液的圆二色谱图;c为该荧光碳量子点的xps全光谱;d为该荧光碳量子点水溶液的光致发光发射光谱,激发波长从340nm逐渐延长到500nm,增量为10nm;插图展示了对发射光谱强度进行归一化的结果;e为激发波长为340nm、发射波长为410nm的条件下,该荧光碳量子点水溶液暴露20分钟或15小时的光致发光强度变化图。
图2是实施例1的附图,是鱼精蛋白修饰的荧光碳量子点的xps高分辨光谱。
图3是实施例1的附图,是bradford法的肽浓度校准曲线,标准物为牛血清白蛋白。
图4是实施例2、3、4的附图,是鱼精蛋白修饰的荧光碳量子点对hek293细胞的细胞毒性评估结果与活细胞成像效果;其中,a、c依次为使用该荧光碳量子点处理hek293细胞3小时后,在激发波长405nm、594nm下的暗场的共聚焦荧光图像,比例尺为40μm;b、d、e依次为使用该荧光碳量子点处理hek293细胞3小时后,在激发波长488nm下的暗场、亮场和重叠图像的共聚焦荧光图像,比例尺为40μm;f为在不同浓度该荧光碳量子点之下暴露24小时后的细胞存活率;g为在激发波长488nm下不同时间的结果图;h为在激发波长405nm下0.5小时的dapi成像图与在激发波长488nm下3小时的该荧光碳量子点对hek293细胞成像图的重叠图像。
图5是实施例4的附图,是鱼精蛋白修饰的荧光碳量子点对hek293细胞的核仁靶向性效果;其中,a为使用rnase处理hek293细胞后,使用该荧光碳量子点处理3小时,得到的暗场、亮场和重叠图像的倒置荧光显微镜图像,比例尺为50μm;b为使用该荧光碳量子点处理hek293细胞3小时后,在激发波长488nm下的三维共聚焦荧光图像,比例尺为15μm;c为该荧光量子碳点结合不同体积rna的滴定曲线。
图6是实施例5、6的附图,展示了该荧光碳量子点与dna纳米复合物的形成以及对hek293细胞的体外转染效果;其中,a为不同质量比(0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2)的荧光碳量子点/pegfp-n1质粒dna的琼脂糖凝胶电泳图谱和质量比为2的tem图像,pdna是指pegfp-n1质粒dna,比例尺为500μm;b为不同质量比(0.5、1、1.5、2、2.5、3)的荧光碳量子点/鲑鱼精dna的琼脂糖凝胶电泳图谱和质量比2的tem图像,dsdna是指鲑鱼精dna,比例尺为200μm;c、d为用质粒与荧光碳量子点、硫酸鱼精蛋白的载体溶液分别形成的纳米复合物处理细胞24小时后,得到的倒置荧光显微镜图像,cd-ptms/pdna是指荧光碳量子点/pegfp-n1质粒dna,ptm/pdna是指硫酸鱼精蛋白/pegfp-n1质粒dna,比例尺为400μm;e-h依次为cd-ptms/pdna、ptm/pdna、cd-ptms、control的转染效率,control是指pei70k/pegfp-n1质粒dna。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
下列实施例中未注明具体实验条件的试验方法,通常按照常规实验条件或按照制造厂所建议的实验条件。所使用的材料、试剂等,如无特殊说明,为从商业途径得到的试剂和材料。
实施例中所述的鱼精蛋白修饰的荧光碳量子点,其包括如下制备步骤:
将20ml乙二醇和溶于12ml超纯水的0.2g硫酸鱼精蛋白溶液混合,涡旋10s以形成均匀系统。转移进50ml锥形瓶,置于家用微波炉中加热(700w加热7.5min)。冷却至室温后,产物全部加入10000mwco透析膜中,用18.2mω·cm的超纯水透析2天,获得澄清淡黄色水溶液约15ml,内含硫酸鱼精蛋白修饰的荧光碳量子点,置于室温保存。
实施例1
本实施例对鱼精蛋白修饰的荧光碳量子点进行性能表征。
在jem-2100f场发射电镜(jeol,美国)上,以200kv的加速电压,用透射电镜(tem)观察了分散在超纯水中的颗粒形貌,所得图像见图1a,该荧光碳量子点呈单分散状,粒径均匀,平均约13nm。
用zetasizernanozs90(马尔文,英国)在超纯水中测量了颗粒的zeta电位,带正电荷,结果为2.99±0.81mv。
使用荧光分光光度计f-7100(日立,日本)测量了光致发光的发射光谱,设定激发波长为340nm,逐渐延长到500nm,所得图谱见图1d。结果表明该荧光碳量子点溶液的发射光谱具有激发波长依赖的特性,覆盖蓝、绿、红光谱范围,最大发射波长为416nm。
使用同样的仪器,进行了光稳定性实验,激发波长为340nm,对该荧光碳量子点溶液连续照射15小时,测定发射波长为410nm处的光致发光强度,所得结果见图1e。连续照射20分钟后,发射强度没有明显损失;即使在照射15小时后,仍保持原强度的80%以上。表明该荧光碳量子点具有优异的抗光漂白能力。
绝对荧光量子产率用c11347quantaurus-qy(滨松光子,日本)测定,结果为2.13%。
在escalab250xispectrometer(赛默飞,美国)上进行了全光谱和高分辨x射线光电子能谱(xps)分析,所得图谱分别见图1c与图2。全光谱表明该荧光碳量子点由c、o、n、s四种元素组成。高分辨率光谱中,c1s谱带显示存在c-c(284.7ev)、c-o/c-n(285.9ev)和c=o(287.8ev)键;n1s谱带显示存在n=c(399.1ev),n-h(399.7ev),n-c(400.2ev);o1s谱带显示存在c=o(531ev)和c-o(532.6ev);s2p谱带表明存在-so3基团,该基团来源于在硫酸存在下的丝氨酸的o-硫酸盐。
在chirascancdspectrometer(appliedphotophysics,英国)上记录了圆二色光谱,所得图谱见图1b。结果表明鱼精蛋白成功连接到碳量子点上,并且在超纯水中呈现统计线圈结构。
为了得到鱼精蛋白在该荧光碳量子点上的蛋白负载率,利用bradford试剂盒(bio-rad,美国)对其中肽进行定量。最大吸收波长为595nm,实验在96孔微孔板(康宁,美国)上进行,分为三组,以牛血清白蛋白(sigma,usa)为标准蛋白,建立标准曲线,见图3。蛋白负载率为67.98±0.9%,计算公式见下:
实施例2
本实施例对鱼精蛋白修饰的荧光碳量子点进行细胞毒性评估。
hek293细胞接种于96孔微孔板中,密度为8000个/孔。培养24小时后,细胞达到80%汇合度,加入该荧光碳量子点浓度为0至40μg/ml的dmem培养基。培养24小时后,采用celltiter-lumitm细胞活力试剂盒(碧云天,中国),使用synergyhtxmulti-modemicroplatereader(biotek,美国)测量样品的发光度。以未暴露细胞(在培养基中)作为对照,细胞活力(平均值%±sd,n=3)表示如下:
所得结果见图4f。该荧光碳量子点在0~35μg/ml剂量下细胞活性高于80%,因此,选择30μg/ml作为后续细胞成像的工作浓度。
实施例3
本实施例对鱼精蛋白修饰的荧光碳量子点进行活细胞核仁的多色成像。
hek293细胞接种于玻璃皿中,密度为12500个/孔。培养24小时后,加入2ml该荧光碳量子点浓度为30μg/ml的无血清dmem,对细胞进行染色。置于fv3000共聚焦激光扫描显微镜(奥林巴斯,日本)的活细胞培养箱中,培养3小时后,设置激发波长为405、488与594nm,采集荧光图像,得到的图像见图4a-e。
位于细胞内的该荧光碳量子点在405、488和594nm激光激发下显示出明显的多色荧光。它们成功抵达并点亮了细胞核的核仁,从而将其与其他细胞区域分开。在实际应用中,发射波长范围可以根据需要进行调整。
实施例4
本实施例对鱼精蛋白修饰的荧光碳量子点进行活细胞核仁的长期实时成像。
细胞染色处理见实施例3,每30分钟采集一次荧光图像,持续12小时。为了进一步证实该荧光碳量子点的核仁靶向性,在染色前,用dapi(索莱宝,中国)处理细胞20分钟,对细胞核进行复染。得到的图像见图4g-h。
细胞对该荧光碳量子点的摄取呈时间依赖性,在每30分钟扫描一次的情况下,在3小时的时间点基本达到恒定,至少可以维持12小时。另外,dapi是一种特异性细胞核染料,在激发波长为405nm有发射,在488nm时无明显发射。值得注意的是,在0.5小时的时间点,该荧光碳量子点很少进入细胞,即此时此激发波长之下,核区只有dapi有成像效果,且点亮了完整的细胞核结构。因此,将此时的dapi成像图与培养3小时、激发波长为488nm的成像图进行融合,两色大部分区域可以叠加,证明该荧光碳量子点可进入细胞核。培养12小时后,该荧光碳量子点仍然在细胞核内发射出强烈的荧光,几乎不发生移位,且细胞没有明显的形态学损伤,因此,它能稳定、实时、长期地(>12h)成像活细胞。
为证明荧光碳量子点核仁靶向性,我们对荧光碳量子点标记的细胞进行了三维成像扫描。如图5b所示,细胞核内明亮的荧光斑点为三维立体结构,表明碳量子点成功对细胞器染色。由于碳量子点进入细胞后大部分富集在细胞核内,因此我们推断碳量子点能特异性地结合细胞核的核仁。由于核仁的主要结构为rrna,因此我们利用rna酶(rnase)对核仁进行了消化实验。
将hek293细胞接种于96孔微孔板中,密度为8000个/孔。培养24小时后,将细胞用4%多聚甲醛固定30分钟,并用50μl的浓度为0.2%的tritonx-100透化8分钟。然后,将这些细胞分为两组,一组加入50μl的浓度为75μg/ml的rnase进行消化,另一组加入50μl的pbs作为对照。孵育3小时后,所有细胞均加入2ml该荧光碳量子点浓度为30μg/ml的无血清dmem,对细胞染色3小时。最后,使用eclipsets2倒置常规显微镜(尼康,日本)进行成像。在每个步骤之前,将细胞用pbs冲洗两次。得到的图像见图5a。
如图5a所示,rna酶消化后细胞核内的明亮荧光斑点消失了,证明此荧光斑点确实为核仁,荧光碳量子点能对核仁进行结合。我们进一步进行了体外rna滴定实验。
将0.5mg核糖核酸酵母溶解在1ml的ph=8.0的te缓冲液中,制备rna储备液。取600μl的浓度为150μg/ml的荧光碳量子点(cd-ptms)水溶液,利用不同体积rna溶液滴定,每次滴加20μl,混合并孵育5分钟。随后,设置激发波长为488nm,利用荧光分光光度计f-7100测量混合物的荧光,并使用超纯水的荧光发射光谱进行校准。得到的结果见图5c。
图5c结果表明,随着rna加入量增多,荧光碳量子点荧光逐渐增强,表明rna可有效与碳量子点结合并增强其荧光。以上结果证明通过此微波法一步合成的荧光碳量子点具有细胞核仁靶向能力。
实施例5
本实施例对鱼精蛋白修饰的荧光碳量子点的核酸运载能力进行评估。
用质粒提取试剂盒dp-103(天根,北京)提取纯化pegfp-n1质粒(赛维尔生物,武汉),并在-80℃下保存。将该荧光碳量子点溶于超纯水中,得到浓度为1mg/ml的载体溶液,取同体积、不同浓度的pegfp-n1质粒或鲑鱼精dna(sigma,美国)溶液与载体溶液,在旋涡下等体积混合,得到不同重量比的载体/dna纳米复合物。然后,将混合物在室温下孵育30分钟,形成稳定的络合物。
制备一系列重量比不同的该荧光碳量子点/dna纳米复合物,备用。取5μl的纳米复合物溶液与0.5μl的midorigreendirectdnastain(nippongenetics,德国),混合,滴入1%琼脂糖凝胶中。在1x-tbe缓冲液(索莱宝,中国)、110v恒定电压的条件下,采用琼脂糖凝胶电泳系统(bio-rad,美国),运行60分钟。结束后,采用azurec600成像系统(azurebiosystems,美国),在蓝光波长下观察并拍摄dna条带,得到的图像见图6a-b。pegfp-n1质粒dna(pdna)和鲑鱼精dna(dsdna)在载体/dna重量比分别高于0.7和2的孔中被完全阻滞,说明该荧光碳量子点能与这两种dna有效结合,成为一种通用核酸载体。
用透射电镜观察载体/dna纳米复合物的形态,载体与dna的重量比为2,得到的图像见图6a-b,成功形成分散的纳米球,平均直径约为50nm,其大小和重量比满足了体外高效转染的要求。进一步证明,通过调整条件,该荧光碳量子点可以成为一种纳米转染载体,实现体外转染。
实施例6
本实施例对鱼精蛋白修饰的荧光碳量子点进行核酸递送与体外转染。
将hek293细胞接种于24孔微孔板中,密度为8000个/孔,培养24小时。
载体/dna纳米复合物的制备方法见实施例5,dna采用pegfp-n1质粒,载体分别采用该荧光碳量子点、硫酸鱼精蛋白和pei70k(麦克林,中国),载体与dna的重量比采用2:1。
吸出孔板中培养基,加入450μl新鲜无血清dmem培养基,再点入50μl载体/dna纳米复合物溶液(含1.5μg质粒和3μg载体)。孵育4小时后,吸出培养基,用pbs洗涤两次,加入新鲜dmem培养基。再孵育24小时,用eclipsets2荧光倒置显微镜(尼康,日本)观察并拍摄绿色荧光蛋白,得到的图像见图6c-d。
收集孔板中的细胞,用流式细胞仪easycyte6-2l(guava,美国)测定表达了绿色荧光蛋白的细胞数占总细胞数的比例,得到的图谱见图6e-h。
综合以上结果,可见该荧光碳量子点相较单纯的鱼精蛋白与传统载体pei,获得了更高的转染效率,是一种更好的纳米转染载体。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!