一种N,N’-亚甲基双丙烯酰胺的合成方法与流程
一种n,n
’‑
亚甲基双丙烯酰胺的合成方法
技术领域
1.本发明涉及一种n,n
’‑
亚甲基双丙烯酰胺的合成方法。
背景技术:
2.n,n
’‑
亚甲基双丙烯酰胺是一种应用很广泛的精细化学品,其分子结构中含有两个可聚合的双键活性基团,具有交联剂单体的特性,可以与多种乙烯类、丙烯酸与丙烯酰类单体共聚得到多种共聚物,由于其拥有两个双键活性基团,有利于在共聚物中有效调控高分子链的结构从而调控材料宏观的物理、化学性能。
3.例如,淀粉和丙烯酰胺以n,n
’‑
亚甲基双丙烯酰胺为交联剂可得到吸水性极佳的高性能吸水树脂,被广泛的应用于土壤保水剂;利用n,n
’‑
亚甲基双丙烯酰胺与丙烯酰胺、丙烯酸丁酯等共聚可得到全水相的涂料印花增稠剂,是实施全水相涂料印花工艺中的关键助剂。
4.目前,n,n
’‑
亚甲基双丙烯酰胺与丙烯酰胺等活性单体交联而成的聚合体被广泛的应用于水利、电力工程、地下工程的建造中,是效果最好、质量最可靠的防水堵漏灌浆材料;用于采油工艺中的压裂液与堵水剂,是石油、建筑行业非常重要的化工原料。近年来,由于n,n
’‑
亚甲基双丙烯酰胺所具有的多官能团聚合位点,也常用于光敏塑料、尼龙感光板、各种胶片和磁带制造、以及新型高级耐高温防火玻璃等生产行业中,是一种多用途、质量可靠的交联剂与化工助剂。国外对n,n
’‑
亚甲基双丙烯酰胺的研究较为成熟,国内对其应用开发和制备方法的研究还较为有限,不少应用领域还有待进一步的研究和开发,从而发挥n,n
’‑
亚甲基双丙烯酰胺更大的潜力和价值。
5.现有合成n,n
’‑
亚甲基双丙烯酰胺的方法公开如下:
6.(1)授权公告号为cn101462979b的中国专利,公开了以丙烯酰胺和甲醛溶液为原料,使用盐酸或硫酸作为催化剂,制备了n,n
’‑
亚甲基双丙烯酰胺。该方法使用无机酸作为催化剂,提高了反应的产率。但是反应过程中需要严格控制体系中反应物的浓度,以避免无机酸催化剂失活。同时产物后处理步骤较多,生产设备较为复杂,且使用的甲醛溶液挥发性较强,在加料过程中生产环境不够友好,工业化生产具有一定的局限性。
7.(2)2008年,martin等人在journal of controlled release中报道了一种利用丙烯酰氯与亚甲基二胺在甲苯中使用碳酸钾作为缚酸剂合成n,n
’‑
亚甲基双丙烯酰胺的方法。该方法可以在有机体系内进行反应,所得产物纯度较高,产率达到90%以上。但是,其原料丙烯酰氯极易遇水分解,并产生大量氯化氢,使得操作较难实现;反应原料亚甲基二胺价格昂贵,反应需在低温、无水条件下进行,反应环境苛刻,生产成本高,不具备工业化基础。
8.(3)2009年,朱露山、项东升等人分别在《应用化工》与《化工中间体》上报道了利用丙烯腈与甲醛使用酸性离子液体催化合成了n,n
’‑
亚甲基双丙烯酰胺。该方法使用了1
‑
甲基
‑3‑
[α
‑
甲基
‑
(4
‑
磺酸苄基)]咪唑硫酸氢盐酸性离子液体作为反应催化剂,催化效率较无机酸大大增强,并且反应后处理较为容易,无需加入大量碱液中和,减少了副产物的生成。但是所使用的酸性离子液体制备过程复杂,价格昂贵,重复循环利用率低,限制了其产业化
应用。
[0009]
综上,现有技术中使用的n,n
’‑
亚甲基双丙烯酰胺普遍存在生产成本高、生产工艺对环境有污染的困境,并且产品中游离甲醛含量较高。
技术实现要素:
[0010]
本发明所要解决的第一个技术问题是针对现有技术的现状,提供一种能降低反应温度、提高产率、减少副产物产生及废液排放的n,n
’‑
亚甲基双丙烯酰胺的合成方法。
[0011]
本发明所要解决的第二个技术问题是针对现有技术的现状,提供一种反应条件温和、容易控制、安全性高、后处理简单、产品纯度高、易于工业化生产的n,n
’‑
亚甲基双丙烯酰胺的合成方法。
[0012]
本发明解决上述技术问题所采用的技术方案为:一种n,n
’‑
亚甲基双丙烯酰胺的合成方法,其特征在于包括以下步骤:
[0013]
(1)将固相多孔材料放入圆底烧瓶中,加入杂多酸的水溶液,加热搅拌回流一段时间后,静置冷却,然后将固相多孔材料滤出,经烘干后得到负载型杂多酸催化剂;
[0014]
(2)以丙烯酰胺和多聚甲醛为原料,加入少量阻聚剂,在酰胺活化试剂及步骤(1)制得的负载型杂多酸催化剂的催化下,进行反应,
[0015][0016]
(3)待步骤(2)反应结束后,先用热水稀释反应体系,再用纱布滤除负载型杂多酸催化剂;所得滤液真空浓缩后转移至结晶釜内,缓慢降温结晶,离心分离得到n,n
’‑
亚甲基双丙烯酰胺晶体,即为目标产物;离心母液中含有酰胺活化试剂,可浓缩继续回用。目标产物经核磁共振氢谱与红外光谱确定结构,产率约87%。
[0017]
优选地,步骤(1)中,所述的固相多孔材料选自活性炭、二氧化硅、mcm
‑
41分子筛中的一种或多种;所述的杂多酸选自磷钨酸、磷钼酸、磷钨酸铯、硅钨酸钠中一种或多种,杂多酸水溶液的质量浓度为5%~15%。
[0018]
优选地,步骤(1)中,回流时间为4h~6h,冷却静置时间为12h~24h;烘干温度为110~120℃,烘干时间为2~6h。
[0019]
优选地,步骤(2)的具体步骤为:将步骤(1)中制备的负载型杂多酸催化剂与酰胺活化试剂置于反应釜内,加入水作为反应溶剂;然后按比例分别加入反应物丙烯酰胺、多聚甲醛、阻聚剂,将体系加热至反应温度,反应一段时间,待有白色固体析出后,继续保温熟料一段时间至反应完全。
[0020]
优选地,步骤(2)中,所述的酰胺活化试剂选自1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐、n
‑
羟基琥珀酰亚胺、n
‑
羟基硫代琥珀酰亚胺、4
‑
二甲氨基吡啶中的一种或两种;所述阻聚剂为对甲氧基苯酚、对羟基苯酚、酚噻嗪中的任意一种。
[0021]
优选地,步骤(2)中,所述负载型杂多酸催化剂的加入量为丙烯酰胺的6wt%
‑
15wt%。
[0022]
优选地,步骤(2)中,所述丙烯酰胺与酰胺活化试剂的摩尔比为1:(0.05~0.25),丙烯酰胺与水的摩尔比为1:(1.2~3.0),丙烯酰胺与多聚甲醛摩尔比为1:(0.92~0.98),
丙烯酰胺与阻聚剂的摩尔比为1:(0.005~0.01)。
[0023]
优选地,步骤(2)中,反应温度为60~85℃,反应时间为0.5h~3h,保温熟料温度为45~75℃,保温熟料时间为1h~4h。
[0024]
优选地,步骤(3)中,加入稀释热水的温度为50~80℃,滤液真空浓缩的真空度为50
‑
100mmhg,滤液真空浓缩的温度为45~60℃,滤液真空浓缩至原滤液体积的1/2~2/3。
[0025]
优选地,步骤(3)中,冷却结晶温度为5~10℃,冷却结晶时间为12h~36h。
[0026]
与现有技术相比,本发明的优点在于:本发明以丙烯酰胺和多聚甲醛为反应原料,采用负载型杂多酸催化剂与酰胺活化试剂协同催化,制备了n,n
’‑
亚甲基双丙烯酰胺;负载型杂多酸催化剂与酰胺活化试剂的协同使用可在不影响杂多酸催化的情况下,与丙烯酰胺中的羰基相互作用,破坏酰胺键的p
‑
π共轭体系,提高氨基的亲核性,增强氨基与多聚甲醛的反应活性,大大提高该反应的选择性,从而在不添加大量的水作为反应溶剂的情况下实现原料的高效转化,提高反应产率。
[0027]
本发明使用的杂多酸催化剂固相负载,反应均在固体催化剂表面进行,体系中无大量的酸性溶剂存在,在后处理过程中无需加入碱液中和,简化了后处理步骤并减少了废水的排放;且由于负载型杂多酸的催化活性高,体系反应温度较低,反应时间较短,避免了丙烯酰胺中的双键高温聚合或其它加成副产物的产生,并大大的降低了能耗;另外,本发明制备工艺简单,反应条件温和,制得的产物为结晶化的n,n
’‑
亚甲基双丙烯酰胺,合成过程中基本无三废产生,所使用的负载型杂多酸和酰胺活化试剂经处理后可循环使用,降低了生产成本。
[0028]
综上所述,本发明反应过程条件温和、控制容易、安全性高、可以获得高纯度产品,且后处理简单,易于工业化生产。
附图说明
[0029]
图1为本发明实施例1中n,n
’‑
亚甲基双丙烯酰胺与商业化样品的红外光谱对比图;
[0030]
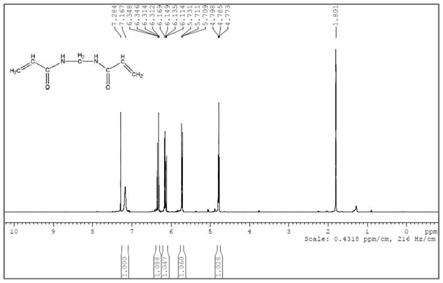
图2为本发明实施例1中n,n
’‑
亚甲基双丙烯酰胺的核磁共振氢谱。
具体实施方式
[0031]
以下结合附图实施例对本发明作进一步详细描述。
[0032]
实施例1:
[0033]
本实施例n,n
’‑
亚甲基双丙烯酰胺的合成方法包括以下步骤:
[0034]
在500l圆底烧瓶中加入250ml、15wt%的磷钨酸溶液,然后加入200g已活化过的二氧化硅微球(直径2.5
‑
4mm),保证二氧化硅微球全部浸入溶液中,加热回流6小时,冷却至室温,静置12小时后,滤出二氧化硅微球,在烘箱中110度烘干3小时,所得催化剂记为cat hpw/sio2;
[0035]
在1l反应釜中,加入60g cat hpw/sio2与1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐191g(1mol),加入250ml去离子水,室温下向体系中加入对甲氧基苯酚7.5g(0.06mol),丙烯酰胺710g(10mol),机械搅拌,分批次向体系中加入多聚甲醛288g(9.6mol)。加料完成后体系温度逐渐升温至75℃,在75℃反应1小时,然后缓慢降温至60℃,
保温熟料2小时至反应完全。然后向体系中加入60℃去离子水150ml,然后趁热将体系中的二氧化硅微球用纱布滤除,二氧化硅微球用60℃少量去离子水(50ml)小心洗涤。所得的滤液小心真空浓缩至约250ml(60℃,80mmhg),倒入结晶釜中,缓慢降温至5℃,静置结晶24小时。离心分离体系中的晶体,即得到目标产物,如图1、2所示,为本实施例所得样品的验证图谱。
[0036]
将上述分离出的晶体室温风干得到晶体1340g,所得晶体采出率为87.18%。
[0037]
实施例2:
[0038]
本实施例n,n
’‑
亚甲基双丙烯酰胺的合成方法包括以下步骤:
[0039]
在500ml圆底烧瓶中加入250ml,6wt%的磷钼酸溶液,然后加入约200g已活化过的mcm
‑
41分子筛,保证分子筛全部浸入溶液中,加热回流4小时,冷却至室温,静置12小时后,滤出分子筛,在烘箱中110度烘干3小时。所得催化剂记为cat hpm/mcm。
[0040]
在250ml圆底烧瓶中,加入20g cat hpm/mcm与1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐38g(0.2mol),4
‑
二甲氨基吡啶24g(0.2mol),加入200ml去离子水,向体系中加入对苯二酚2.2g(0.02mol),丙烯酰胺287g(4mol),室温机械搅拌,分批次向体系中加入多聚甲醛117g(3.92mol)。加料完成后体系温度逐渐升温至75℃,在75℃反应0.5小时,然后缓慢降温至65℃,保温熟料2小时至反应完全。然后向体系中加入65℃去离子水150ml,然后趁热将体系中的二氧化硅微球用纱布滤除,二氧化硅微球用65℃少量去离子水(约25ml)小心洗涤。所得的滤液小心真空浓缩至约200ml(60℃,80mmhg),倒入结晶釜中,缓慢降温至5℃,静置结晶24小时。离心分离体系中的晶体,室温风干,得到晶体540g,所得晶体采出率为87.54%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1